Introduction:
Cystic fibrosis (CF) is a hereditary condition that impacts the respiratory, digestive, and reproductive systems. It is caused by a mutation in the gene that codes for a protein called cystic fibrosis transmembrane conductance regulator (CFTR). This protein regulates the movement of salt and water in and out of cells, and is involved in the function of many organs and tissues in the body.
Frequency:
Cystic fibrosis (CF) is a rare genetic disorder that affects the respiratory, digestive, and reproductive systems. It occurs in about 1 in every 3,000 live births in the United States, and it is one of the most common life-shortening genetic disorders in the country. CF is more common in people of Northern European ancestry, but it can affect people of any race or ethnicity.
Causes:
It is caused by mutations in the gene that codes for the cystic fibrosis transmembrane conductance regulator (CFTR) protein. This protein plays a critical role in regulating the transport of salt and water in and out of cells, particularly in the mucus-secreting glands of the respiratory and digestive tracts.
CF is an autosomal recessive disorder, which means that a person must inherit two copies of the mutated CFTR gene (one from each parent) to develop the condition. If a person inherits one mutated gene and one normal gene, they are called carriers. Carriers do not have CF, but they can pass the mutated gene on to their children.
Mutations in the CFTR gene can affect the normal production or function of the CFTR protein found in cells of the lungs and other parts of the body in persons with CF.
A build-up of thick mucus results from mutations in the CFTR gene that impair or prevent the production of the CFTR protein, which in turn causes recurrent lung infections, pancreatic damage, and difficulties in other organs.
There are more than 1,700 known mutations of the CFTR gene, and the severity of CF can vary widely depending on the specific mutation a person has. Some mutations cause a complete absence of CFTR function, while others cause reduced function. The severity of CF is also influenced by environmental factors, such as air pollution and infections, which can exacerbate the symptoms of the disorder.
Protein production, protein processing, gating, conduction, and inadequate protein are the five main types of CFTR mutations.
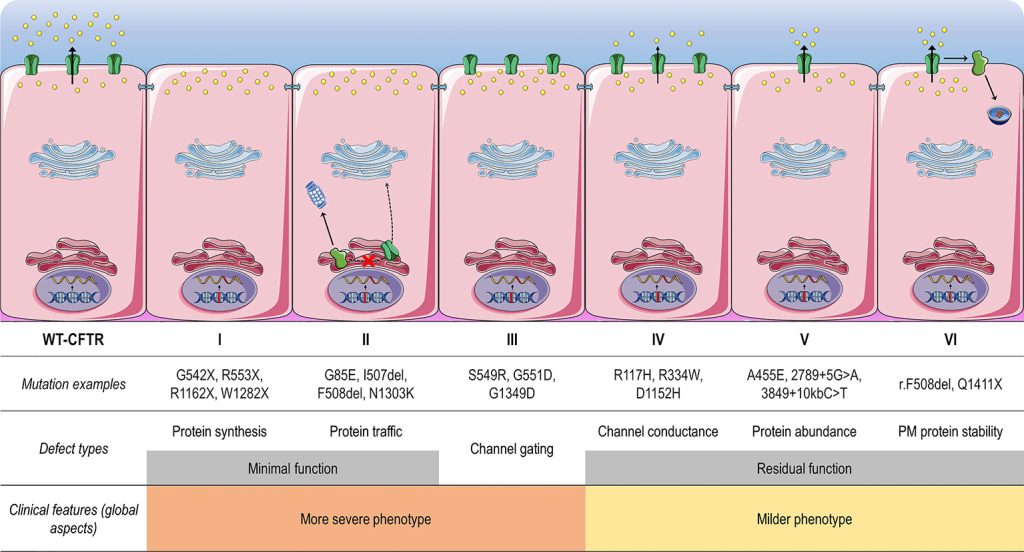
Fig: Classes of CF transmembrane conductance regulator (CFTR) mutations
The CFTR gene is 230,000 base pairs long and produces a 1,480 amino acid protein. It is located on chromosome 7 at the q31.2 locus. The most prevalent mutation, F508, is a three-nucleotide deletion that leads in the amino acid phenylalanine (F) being lost at the 508th position on the protein. This mutation is responsible for two-thirds of all CF cases globally (66-70%).
The most prevalent CF mutation, F508del, is thought to be a protein processing problem classified as a class II defect. The F508del mutation alters the CFTR protein by removing one amino acid.
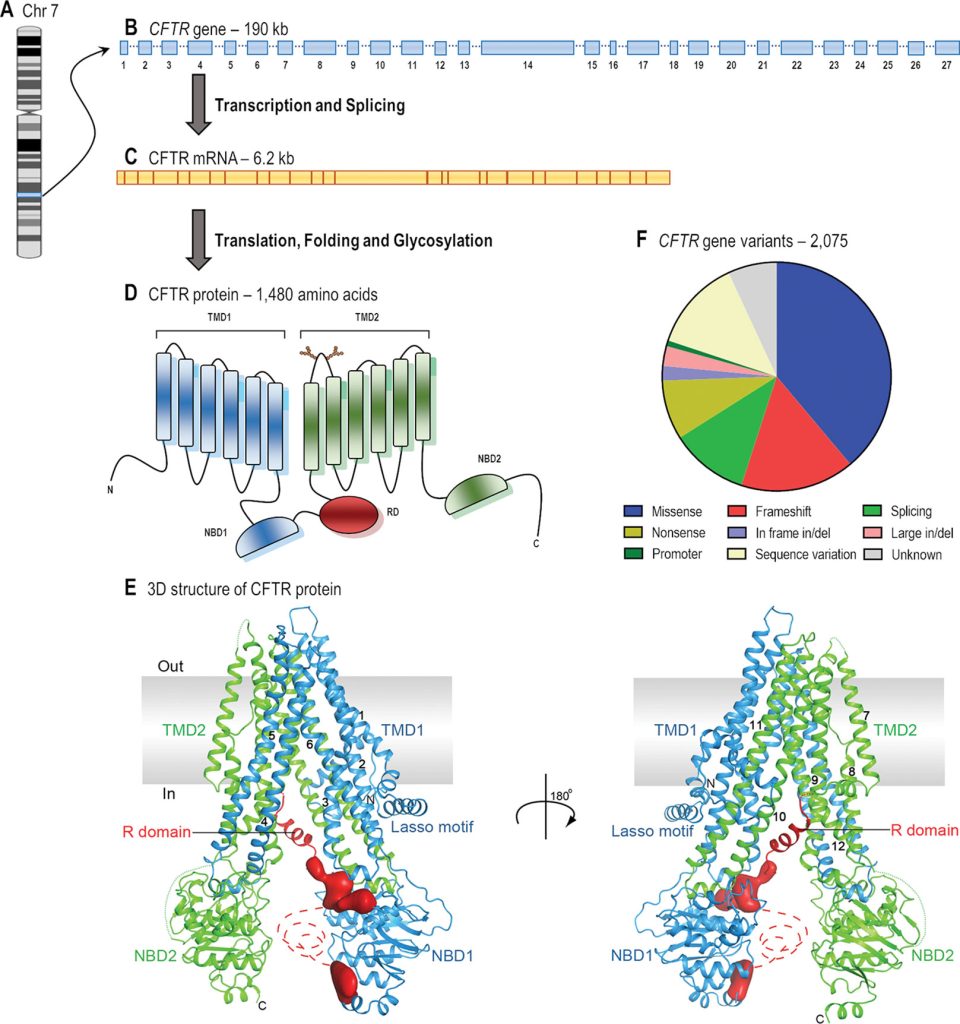
Fig: From gene to protein structure of CFTR gene
CF is most common in people of Northern European ancestry, but it can affect people of any race or ethnicity. It occurs in about 1 in every 3,000 live births in the United States, and it is one of the most common life-shortening genetic disorders in the country.
Sign and Symptoms:
The signs and symptoms of cystic fibrosis (CF) can vary widely, depending on the severity of the condition and the specific mutations a person has in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. It is important to note that not all people with CF will have all of these symptoms, and the severity of the symptoms can vary widely from person to person. Some people with CF may have very mild symptoms, while others may have severe symptoms that require ongoing medical treatment.
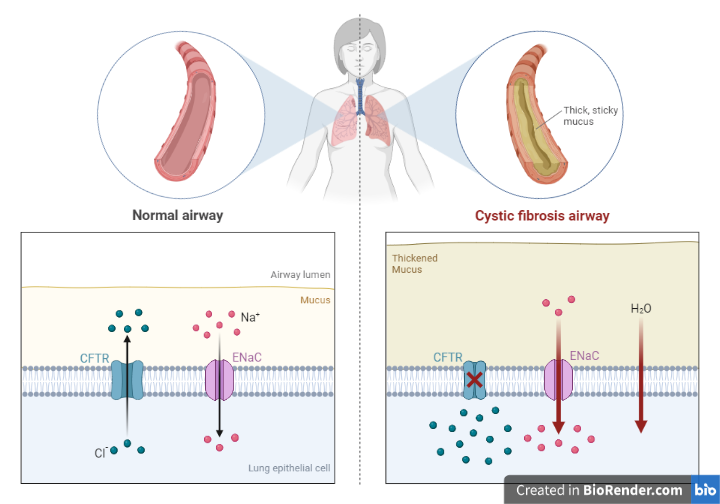
Fig: Cystic Fibrosis Airways
- Respiratory system: CF can cause the airways to become clogged with thick, sticky mucus, leading to persistent coughing, wheezing, and difficulty breathing. People with CF are also at increased risk of lung infections, such as pneumonia or bronchitis.
- Digestive system: CF can cause problems with the absorption of nutrients from food, leading to malnutrition and poor growth or weight gain despite a good appetite. It can also cause frequent, greasy, bulky stools and difficulty with bowel movements. In some cases, CF can cause a build-up of thick, sticky mucus in the pancreas, which can lead to digestive problems.
- Reproductive system: In men with CF, the thick mucus can block the tubes that transport sperm, leading to infertility. Women with CF may have irregular or absent periods, and they may also have fertility problems.
- Sweat glands: CF can affect the sweat glands, leading to excessive sweating and salt loss.
- Liver: Some people with CF may develop liver problems, such as bile duct abnormalities or cirrhosis.
Diagnosis:
Cystic fibrosis (CF) can be difficult to diagnose because the signs and symptoms can vary widely from person to person and may be similar to those of other respiratory and digestive disorders. The diagnosis of CF often involves a combination of tests and evaluations, including:
It is always significant to note that the diagnosis of CF can be complex and may require multiple tests and evaluations. It is also important to remember that not all people with CF will have the same signs and symptoms, and the severity of the symptoms can vary widely from person to person.
- Physical examination: A healthcare provider will perform a physical examination to look for signs and symptoms of CF, such as persistent coughing, wheezing, and difficulty breathing. They may also check for clubbing (enlargement) of the fingertips and nails and assess the person’s growth and weight.
- Sweat test: The sweat test is the most common and reliable test for CF. It measures the amount of salt in a person’s sweat. People with CF have higher levels of salt in their sweat because of problems with the transport of salt and water in and out of cells.
- Genetic testing: Genetic testing can be used to identify mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that cause CF. This type of testing is usually done on a sample of the person’s blood or saliva.
- Other tests: Other tests that may be used to diagnose CF include lung function tests, imaging tests (such as chest X-rays or CT scans), and tests to assess the person’s nutritional status and the functioning of the pancreas.
Treatments:
There is no cure for cystic fibrosis (CF), but there are treatments available that can help manage the symptoms and improve the quality of life for people with the condition. The specific treatment plan for a person with CF will depend on the severity of their condition and the specific symptoms they are experiencing.
Medications: People with CF may be prescribed medications to help clear mucus from the airways, reduce inflammation, and prevent or treat infections. These may include bronchodilators, inhaled steroids, antibiotics, and mucolytics (medications that help thin and loosen mucus).
Chest physical therapy: Chest physical therapy involves tapping and patting the chest and back to help loosen and clear mucus from the airways. It is usually done with the help of a therapist or at home with a parent or caregiver.
Airway clearance devices: There are several devices available that can help clear mucus from the airways, including vibrating vest systems, positive expiratory pressure (PEP) masks, and high-frequency chest wall oscillation (HFCWO) devices.
Nutritional support: People with CF may have difficulty absorbing nutrients from food, so they may need to take supplements or receive nutrition through a feeding tube.
Lung transplant: In severe cases, a lung transplant may be an option. However, this is a major surgery with significant risks and is not suitable for all people with CF.
References:
- https://my.clevelandclinic.org/
- https://med.stanford.edu/
- https://www.mayoclinic.org/
- https://www.cff.org/
- Davis, P.B., 2006. Cystic fibrosis since 1938. American journal of respiratory and critical care medicine, 173(5), pp.475-482.
- Bell, S.C., Mall, M.A., Gutierrez, H., Macek, M., Madge, S., Davies, J.C., Burgel, P.R., Tullis, E., Castaños, C., Castellani, C. and Byrnes, C.A., 2020. The future of cystic fibrosis care: a global perspective. The Lancet Respiratory Medicine, 8(1), pp.65-124.
- Lopes-Pacheco, M., 2020. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Frontiers in pharmacology, 10, p.1662.