Introduction to Tight Junctions:
The epithelium and endothelium segregate the tissue compartments in a host body, and the host is protected from the outside world. Tight junctions selectively control what gets into and out of the cell. Tight Junctions are the intercellular contacts that prevent harmful entities present in the lumen of the stomach from invading the gastric mucosa.
Structure and Components of Tight Junctions:
Tight junctions’ integrity is maintained by three components, viz, transmembrane proteins (Occludin, Claudins, Junctional Adhesion Molecules [JAM], and tricellulin), cytoskeletal elements, and cytoplasmic scaffolding proteins (Zonulae).
Mechanism of Tight Junction Disruption by Helicobacter pylori:
Overview of H. pylori Pathogenesis
Helicobacter pylori is a helical, gram-negative bacterium that invades the gastric lining of the stomach and forms dense colonies along the luminal surface of the mucous-secreting cells. It often localizes near the tight junctions between these cells and can exploit these junctions to penetrate the mucosal layer and access the underlying lamina propria. H. pylori has a propensity towards the epithelial cells that express Limax flavus agglutinin.
H. pylori is evident to colonize the gastric epithelium of approximately 50% of the world’s population. H. pylori compromises the connections between epithelial cells and weakens the integrity of the stomach’s gastric barrier.
Cag Pathogenicity Island and CagA Protein Function
Strains associated with gastritis typically carry the cagpathogenicity island (cag PAI), a key virulence factor. Cag PAI makes effectors of a bacterial type IV secretion system and exports CagA, a product of cag PAI, across the bacterial membrane and into the epithelial cells of the host’s stomach.
CagL, a protein found on the pili of H. pylori’s type IV secretion system, can bind to the α5β1 integrin on host cells. This secretion system may also trigger the movement of phosphatidylserine from inside to the outside of the host cell membrane, allowing the CagA protein to attach to it and enter the epithelial cells. Once inside host cells, CagA undergoes tyrosine phosphorylation at specific EPIYA sequences, leading to structural changes that promote cell movement and may weaken the stomach’s protective lining. Even without phosphorylation, CagA influences gastric epithelial cells by activating β-catenin, disturbing junctional complexes, and disrupting cell polarity. It also binds to E-cadherin and other adaptor proteins, interfering with cell-to-cell adhesion and polarity. Additionally, CagA affects tight junction integrity by interacting with PAR1 b, a kinase that regulates epithelial polarity by modifying microtubule-associated proteins and destabilizing microtubules to support polarized molecule distribution. This interaction ultimately causes ZO-1, a protein normally confined to the cell junctions, to become unevenly distributed, resulting in fragmented staining along the cell edges.
Effect on Occludin, Claudins, ZO-1, and E-cadherin
Claudins that regulate paracellular permeability (the movement of substances across an epithelial barrier through the spaces between the cells, rather than through the cells themselves) are likely the target of inflammation or H. pylori itself. Research has demonstrated that H. pylori interferes with the proper positioning of occludin at tight junctions. Occludin plays a key role in controlling the permeability and functional integrity of the gastric epithelial barrier.
Dysregulation of the adherens junction by H. pylori can also be deployed for invasion by H. pylori-translocated CagA that interacts with E-cadherin and p120. This destabilizes the adherens junction.
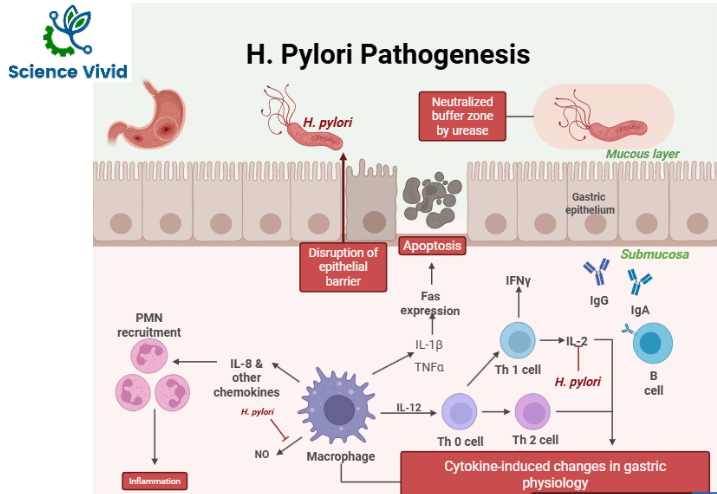
Fig: Pathogenesis mechanisms of H. pylori
Other Pathogens Causing Tight Junction Disruption
Enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC) inject effector molecules into host epithelial cells by a type 3 secretion system (T3SS) to hijack the host cell. After the invasion of EPEC into host epithelial cells, the paracellular permeability of the intestinal epithelium is altered, leading to diarrhea. EPEC is studied to compromise Occludin, Claudin 1, and ZO-1, leading to progressive loss of Transepithelial Electrical Resistance (TER). EHEC lowers TER, alters ZO-1, causes actin rearrangement, inhibits the renewal of tight junction proteins, and increases paracellular flux of mannitol, leading to diarrhea.
The tight junction transmembrane proteins claudin-3 and -4 are the binding sites of bacterial toxin Clostridium perfringens enterotoxin (CPE). Exposure of intestinal cells to CPE results in tissue damage followed by fluid and electrolyte secretion. After binding to the cell surface, CPE remains associated with the plasma membrane, forms pores in the plasma membrane, and increases membrane permeability.
Diarrheogenic strains of Bacteroides fragilis produce a B. fragilis enterotoxin (BFT) or fragilysin and disrupt tight junctions by proteolytic degradation of tight junction proteins, including actin. In an experiment, it caused a decrease in TER and an increase in paracellular permeability.
References:
- Dunne, C., Dolan, B., & Clyne, M. (2014). Factors that mediate colonization of the human stomach by Helicobacter pylori. World journal of gastroenterology, 20(19), 5610–5624. https://doi.org/10.3748/wjg.v20.i19.5610
- Wroblewski, L. E., & Peek, R. M., Jr (2011). “Targeted disruption of the epithelial-barrier by Helicobacter pylori”. Cell communication and signaling : CCS, 9(1), 29. https://doi.org/10.1186/1478-811X-9-29