Introduction:
- Sanger sequencing is a method of DNA sequencing that was developed by Frederick Sanger in the 1970s. It is a chain termination method that uses modified nucleotides, called dideoxynucleotides, to stop the synthesis of a growing DNA strand. The resulting DNA fragments are then separated by size on a gel, and the sequence of the original DNA strand is determined by reading the order of the nucleotides at the end of each fragment.
- Sanger sequencing is relatively inexpensive and widely used for sequencing short stretches of DNA, and it is still used today for some applications, particularly for small genome sequencing projects, and for sequencing PCR products.
- It is considered a first-generation sequencing technology and has been largely replaced by next-generation sequencing techniques such as Illumina, PacBio and Nanopore, which are capable of sequencing much larger amounts of DNA much faster and at a lower cost.
- Sanger sequencing was a major breakthrough in the field of genetics and genomics and has played a crucial role in the development of the field of molecular biology. It was the first method to allow scientists to determine the complete sequences of DNA.
History:
- Sanger sequencing, also known as dideoxy sequencing, is a method of DNA sequencing that was developed by Frederick Sanger in the 1970s. Sanger was awarded a Nobel Prize in Chemistry in 1980 for his work on this method.
- The method was first published in 1977 in two papers, one in Nature and one in the Proceedings of the National Academy of Sciences. Sanger and his colleagues used the method to sequence a 5,386 base pair DNA fragment from the bacteriophage phiX174. This was the first time a complete DNA sequence of an organism had been determined, and it marked the beginning of the modern era of genetics and genomics.
- Sanger sequencing was widely adopted as the standard method for DNA sequencing and was used to sequence the entire genome of the first organism, the bacteriophage phiX174, in 1983. Sanger sequencing was also used to sequence the first eukaryotic genome, the yeast Saccharomyces cerevisiae, in 1996.
- In the late 1990s and early 2000s, Sanger sequencing was largely replaced by next-generation sequencing technologies such as Illumina, PacBio and Nanopore, which are capable of sequencing much larger amounts of DNA much faster and at a lower cost. However, Sanger sequencing is still used today for some applications, particularly for small genome sequencing projects and for sequencing PCR products.
Principle:
The principle of Sanger sequencing is based on the incorporation of modified nucleotides, called dideoxynucleotides, into a growing DNA strand during PCR. Dideoxynucleotides are chemically modified nucleotides that lack the 3′-OH group required for the extension of the DNA strand. These modified nucleotides terminate DNA synthesis when they are incorporated, resulting in a set of DNA fragments of different lengths.
The process begins by creating a set of DNA strands that are identical to the template strand except that they each contain a single dideoxynucleotide at a specific location. This is done by using a primer to initiate DNA synthesis and adding a mixture of the four regular nucleotides (dATP, dCTP, dGTP, dTTP) and the four dideoxynucleotides (ddATP, ddCTP, ddGTP, ddTTP) to the reaction.
When the DNA polymerase encounters a dideoxynucleotide, it incorporates it into the growing strand, but the lack of the 3′-OH group prevents further extension, creating a set of DNA fragments with different lengths. These fragments are then separated by size on a gel, and the sequence of the original DNA strand is determined by reading the order of the nucleotides at the end of each fragment.
Workflow:
The various form of Sanger sequencing protocol exists and some steps can be adjusted depending on the specific application, for example, using different primers, different PCR conditions, and different gel electrophoresis buffer.
Sanger sequencing protocol consists of several steps and the protocol is widely used and well-established, but some variations exist depending on the specific application.
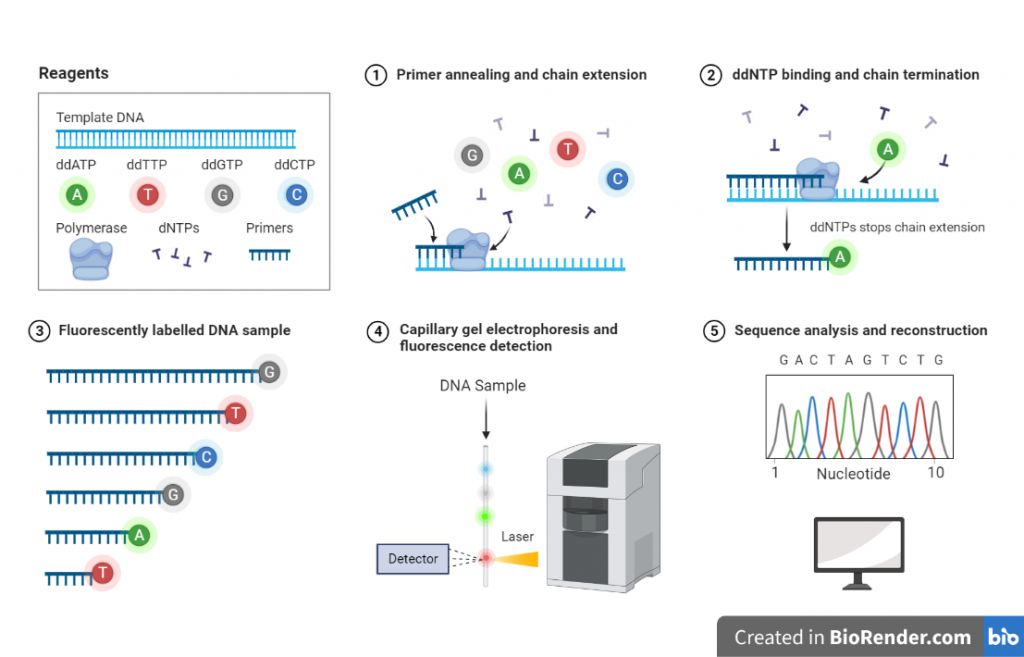
Fig: Workflow of DNA sequencing
Template preparation
The DNA sample to be sequenced is isolated and purified. It can be a plasmid, a genomic DNA or PCR product.
Primer design
A primer is designed that is complementary to the region of the DNA to be sequenced.
PCR amplification
The template DNA is amplified using a primer that is complementary to the region of the DNA to be sequenced. This is done using a thermostable polymerase such as Taq polymerase.
Chain termination
During the PCR amplification, a mixture of the four regular nucleotides and the four dideoxynucleotides is added to the reaction. When the DNA polymerase encounters a dideoxynucleotide, it incorporates it into the growing strand, but the lack of the 3′-OH group prevents further extension, creating a set of DNA fragments with different lengths.
PCR cleanup
PCR cleanup is a method used to purify and separate the amplified DNA product (PCR product) from the remaining components of the PCR reaction mixture. The purpose of PCR cleanup is to remove unincorporated primers, nucleotides, enzymes, and other contaminants that may interfere with downstream applications.
Gel electrophoresis
The resulting DNA fragments are separated by size on a gel using electrophoresis. Typically, the DNA fragments are loaded on an agarose gel and run in a buffer of Tris-Acetate-EDTA (TAE) or Tris-Borate-EDTA (TBE).
Sequence detection and analysis
The order of the nucleotides at the end of each fragment is determined by a process called sequence detection and analysis. This is typically done using a fluorescent dye that binds to the nucleotides during the PCR amplification and is detected using a laser. The resulting data is then analyzed to determine the sequence of the original DNA strand.
Sequencing cleanup
This step of DNA sequencing reactions removes unincorporated BigDye terminators and salts. Sequencing cleanup is a method used to purify and separate the sequencing products (DNA fragments) from the remaining components of the sequencing reaction mixture. The purpose of sequencing cleanup is to remove unincorporated nucleotides, enzymes, and other contaminants that may interfere with downstream applications, such as sequencing or cloning to obtain accurate and reliable results.
Data analysis
The sequence data is then analyzed and compared to known sequences in databases to confirm the identity of the sample.
Trace file analysis: The first step in data analysis is to view the trace files, which are the output files generated by the sequencing instrument. The trace files contain information about the intensity of the fluorescent signals at each base position, as well as the electrophoresis trace, which shows the mobility of the DNA fragments during the sequencing reaction.
Base calling: The second step is to assign the base calls, which is the process of determining the nucleotide at each position in the sequence. This is typically done using software that analyzes the trace files and makes predictions about the nucleotide at each position based on the intensity of the fluorescent signals.
Quality control: The third step is to check the quality of the sequencing data. This includes checking for sequence errors, such as base calling errors and sequencing artifacts, as well as assessing the quality of the sequences by calculating various quality metrics, such as the Phred quality score.
Assembly: The fourth step is to assemble the sequences generated from different reactions to obtain the final sequence. This step is only needed if the DNA fragment to be sequenced is too long to be sequenced in one reaction.
Annotation: The final step is to annotate the sequence, which involves adding information about the features of the sequence, such as exon-intron boundaries, promoter regions, and the location of coding regions.
Instruments:
ABI 3730 DNA Analyzer: This is a capillary electrophoresis-based instrument that is widely used for Sanger sequencing and genotyping applications. It can sequence up to 96 samples in parallel and has a high throughput.
Li-Cor 4200 DNA Analyzer: This is a capillary electrophoresis-based instrument that uses infrared dye-labeled primers for Sanger sequencing.
MegaBACE 1000 DNA Sequencer: This is a gel-based instrument that uses a laser to detect the fluorescent dyes incorporated during Sanger sequencing. It can be used for both DNA sequencing and fragment analysis.
CEQ 8000 Genetic Analysis System: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Applied Biosystems 3130 Genetic Analyzer: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Beckman Coulter CEQ 8000: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Genetic Analyzer 3500: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Genetic Analyzer 3730/3730xl: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Genetic Analyzer 3500xL: This is a capillary electrophoresis-based instrument that is commonly used for Sanger sequencing and genotyping applications.
Limitations:
- Sanger sequencing can only sequence one fragment at a time and requires a separate reaction for each fragment. This makes it time-consuming and expensive to sequence large genomes or multiple samples.
- Sanger sequencing has a relatively high error rate, especially at low signal intensities. This can make it difficult to obtain accurate and reliable results, particularly for low-complexity regions of the genome.
- Sanger techniques can only sequence small segments of DNA (between 300 and 1000 base pairs).
- Because the primer binds in the first 15 to 40 bases of a Sanger sequence, the quality of the sequence is frequently poor.
- The quality of sequence degrades after 700 to 900 bases.
- Sanger sequencing cannot be properly quantified, but can detect mosaic mutations in as few as 20% of cells.
- Sanger sequencing is less sensitive than many other DNA sequencing methods, making it difficult to detect rare or low-abundance mutations.
Sanger sequencing vs. next-generation sequencing:
- Scale: Sanger sequencing is capable of sequencing a single DNA fragment of up to 1000 base pairs, while next-generation sequencing can sequence billions of base pairs at once.
- Speed: Sanger sequencing is a slower process, taking several days to complete a single sequencing run, while next-generation sequencing can produce results in a matter of hours.
- Cost: Sanger sequencing is more expensive than next-generation sequencing, due to the need for large amounts of reagents and the use of radioactive materials.
- Throughput: Sanger sequencing can only sequence one DNA fragment at a time, while next-generation sequencing can sequence multiple samples in parallel, increasing the throughput and decreasing the cost per sample.
- Applications: Sanger sequencing is mostly used for smaller projects such as sequencing a gene, while next-generation sequencing is used for larger projects such as genome sequencing and transcriptome analysis.
- Read length: Sanger sequencing has a longer read length than next-generation sequencing, which is usually around 25-300 bp, while NGS has read length between 75-150 bp.
- Error rate: Sanger sequencing has a lower error rate than next-generation sequencing, but next-generation sequencing has a higher read coverage and accuracy
Applications:
Diagnostics: Sanger sequencing is used to detect mutations in genes associated with inherited diseases, such as cystic fibrosis and sickle cell anemia. Sanger sequencing is employed to sequences the specific regions of the HLA genes to determine the specific human leukocyte antigen (HLA) type of an individual.
Identification of genetic variations: Sanger sequencing is used to identify genetic variations that may be associated with certain diseases or conditions, such as cancer or psychiatric disorders.
Genetic mapping: Sanger sequencing is used to map the locations of genes on chromosomes, which can provide insights into the genetics of complex diseases and inherited traits.
Gene cloning: Sanger sequencing is used to confirm the sequence of a cloned gene, and to ensure that it has been accurately and completely copied.
Bacterial and viral identification: Sanger sequencing can be used to identify bacterial and viral pathogens by sequencing the conserved regions of their genomes.
Protein engineering: Sanger sequencing is used to determine the sequence of proteins, which is important for understanding their function and for designing new proteins with specific properties.
Identifying genetic variations in population studies: Sanger sequencing can be used to identify genetic variations in population studies, which can provide insights into the genetic basis of disease and population history.
References:
Next Generation DNA Sequencing: A Review of the Cost Effectiveness and Guidelines, https://www.ncbi.nlm.nih.gov/books/NBK274079/
Alicia Gomes, Bruce Korf, Chapter 5 – Genetic Testing Techniques, Editor(s): Nathaniel H. Robin, Meagan B. Farmer, Pediatric Cancer Genetics, Elsevier, 2018, Pages 47-64,