
Introduction:
Agarose gels are a standard component of gel electrophoresis, an effective technique used in the separation and analysis of Deoxyribonucleic acid (DNA) fragments. Because most DNA molecules and fragments that are commonly analysed are significantly larger than proteins, and it would be unable to penetrate a polyacrylamide gel, an agarose gel with a higher pore size is recommended.
Principle:
The mobility of a charged molecule in a medium subjected to an electric field is the fundamental separating principle in all electrophoresis.
v=Eq/f
where,
V denotes the electrophoresis molecule’s velocity.
The electrical field is measured in volts/cm, the net charge on the molecule is q, and the frictional coefficient is f. The influence of f is affected by the molecule’s mass and shape.
This equation simply states that a particle’s rate (v) is affected by the electrical field and charge, but is influenced inversely by the counteracting force caused by resistance. The size and form of the molecule are, of course, factors that influence F.
Components/ Instrumentation:
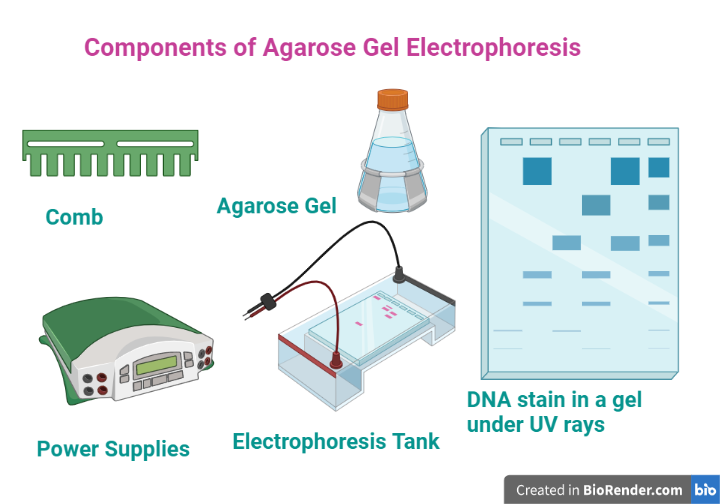
Fig: Components of Agarose Gel Electrophoresis
Agarose
Agarose is a natural polymer composed of D-galactose and 3,6-anhydro-L-galactose repeating units and is derived from seaweed (red algae). It’s a polysaccharide that’s utilized in agarose gel electrophoresis to separate nucleic acids by size. It generates an inert matrix that is widely utilized in biochemistry and molecular biology separation techniques such as electrophoresis and chromatography.

Fig: Chemical structure of agarose
Agarose comes in five different forms for different applications: Low EEO (Electroendosmosis), Medium EEO, High EEO, Special High EEO, and Quick Soluble (QS).
Agarose LE is indicated for cloning investigations and is best for analysing nucleic acids from 50 bp to 25 kbp. For nucleotide analysis below 1 kb, high resolution agarose is preferable. Low melting (LM) agarose makes it easier to separate and purify big intact DNA fragments; it’s ideal for applications that need enzymatic processing or cloning.
A table describing the separation of DNA is shown below.

Fig: A table describing the separation of DNA
Features
- Extraordinary mechanical resistance makes handling more trustworthy and simpler.
- The gel has excellent visibility and clarity.
- Staining agents are absorbed very slowly.
- Modifying the gel concentration allows for pore size to be adjusted in accordance with particle size.
- Absence of toxicity.
- It’s perfect for resolving DNA and RNA fragments with sizes ranging from 100 bp to >30 kb.
- In routine applications, it’s ideal for analysing and recovering DNA and RNA.
- Better handling and fewer breakage are made possible by the gel’s strong structure.
- Ouchterlony (antigen-antibody interaction assay) and radial immunodiffusion (RID) are two examples of protein electrophoresis applications (antigen quantitation assay).
- Furthermore, the performance of agarose is considered in terms of morphology, moisture, gel strength, melting temperature, sulphate content, electroendosmosis, and DNase/RNase activity to ensure that stated parameters are achieved.
Electrophoresis buffer
Depending on the application, either Tris-Acetate-EDTA (TAE) or Tris-Borate-EDTA (TBE) buffer can be used to prepare and run the gel. The TAE buffer helps linear DNA to migrate quicker and genomic and supercoiled DNA to be resolved better on electrophoresis. TBE buffers, on the other hand, offer a higher buffering capacity, making them suited for electrophoresis at higher voltages. They are highly recommended when separating < 2 kb fragments However, because borate inhibits enzyme activity, it is not suggested for enzymatic applications.

Fig: Table illustrating an optimal buffer concentration for best nucleic acid fragment separation.
Loading buffer (Gel Loading dye)
To effectively visualize the samples during loading and tracing in electrophoresis, the dyes xylene cyanol FF (XC) and bromophenol blue (BB) plus 30% glycerol in water are frequently used. The glycerol increases the density of the final solutions, causing them to settle to the bottom of the wells.
Sucrose (40%), Ficol (25-30%), or glycerol (30-50%) can all be used as a replacement.
Staining Solution
Ethidium Bromide (EtBr)
The use of ethidium bromide (intercalating agent) to visualize DNA in gels is a simple, sensitive, and reliable method. The EtBr inter-linked to DNA fluoresces brightly under the presence of short or medium wavelength UV light from UV- transilluminator.
EtBr is a mutagen and potential carcinogen because it binds to DNA. EtBr should be handled with care and kept to a minimum. When utilizing EtBr or handling anything that could be contaminated with EtBr, you should always wear gloves and a lab coat (such as gel staining containers).
SYBR Safe DNA Stain
SYBR Safe is a non-mutagenic, cyan-based form of SYBR Green dye. The dye absorbs blue light, fluoresces only when it comes into contact with DNA, and then emits green light (lambda max 520 nm). SYBR Safe is slightly less sensitive than ethidium bromide, but it may be seen with blue light instead of UV light, which prevents DNA damage.
DNase/RNase free water
All molecular biology applications require DNase/RNase-Free Distilled Water. It’s been filtered via a 0.1-m membrane and tested for DNase and RNase activities.
Template sample/Starting material
It is the sample (DNA/RNA/Protein) of our interest that is extracted using a specific DNA extraction or nucleic acid extraction technique, which can be manual, kit-based, or automated, depending upon the requirements.
Molecular ladder/Gene ladder
Molecular weight markers, often known as ladders, are a collection of standards used to estimate the size of a protein or nucleic acid fragment on an electrophoresis gel. A solution containing known-size DNA fragments that can be used to estimate fragment sizes in unknown samples.
Electrophoresis Gel casting trays:
They’re made of UV transparent plastic and available in a number of sizes. While the gel is being cast, the open ends of the trays are taped shut, then removed before electrophoresis.
Sample combs
It is a consumable around which molten medium is poured to form sample wells in the gel.
UV Transilluminator (an ultraviolet light box)
It is generally used to visualize stained DNA in gels.
Factors affecting the rate of migration of sample:
Rate of migration of nucleic acids in agarose gels electrophoresis depends on several
important parameters.
Agarose concentrations
Separating lower molecular weight DNA and RNA fragments requires a higher concentration of gels, and vice versa.
Molecular weight
The molecular weight of a DNA fragment is inversely proportional to its rate of migration.
Conformation
The fastest moving DNA is supercoiled DNA, which is followed by linear forms and relaxed open circular forms.
Applied Voltage
At low voltage (<5V/cm) the rate of migration is directly proportional to the applied
voltage. However, if the voltage is increased, mobility of high molecular weight DNA
fragments increased differentially.
Temperature
In electrophoresis, temperature plays a minor role. The band becomes more solid or neatly packed in gel as the temperature drops.
Methodology:
Preparation of the Gel
In an Erlenmeyer flask, weigh out the appropriate quantity of agarose. The w/v % solution is used to make agarose gels. The amount of agarose in a gel is determined by the size of the DNA fragments to be separated, with most gels ranging from 0.5 to 2%. The buffer volume should not exceed 1/3 of the flask’s capacity.

Figure: Pore formation and temperature-induced state transition in agarose gel
- To the agarose-containing flask, add running buffer. To mix, blend the ingredients together. TAE (40 mM Tris-acetate, 1 mM EDTA) and TBE (40 mM Tris-acetate, 1 mM EDTA) are the most commonly used gel running buffers (45 mM Tris-borate, 1 mM EDTA).
- Heating in a microwave is the most frequent method, however it can also be done over a Bunsen flame. Remove the flask for 30 seconds at a time and spin the contents to thoroughly combine them. Repeat until all of the agarose has dissolved.
- To a concentration of 0.5 g/ml, add ethidium bromide (EtBr). Alternatively, after electrophoresis, the gel can be stained for 15-30 minutes in running buffer containing 0.5 g/ml EtBr, followed by destaining in running buffer for the same amount of time.
Note: Because EtBr is a probable carcinogen, it must be appropriately disposed of according to the guidelines of disposable of wastes. When handling EtBr-containing gels, gloves should always be worn. There are other dyes for staining DNA, but EtBr remains the most preferred due to its sensitivity and low cost.
- Allow the agarose to cool on the counter or on the bench. To create the wells, place the gel tray in the casting device and a suitable comb in the gel casting tray. In the casting tray, pour the melted agarose. Allow the agarose to cool to room temperature before using it. Place the gel in the gel box after removing the comb. Alternatively, cover the gel in plastic wrap and keep it at 4 °C until needed.

Fig: Key steps in agarose gel electrophoresis
Setting up of Gel Apparatus and Separation of DNA Fragments
- Add loading dye to the samples that need to be separated, which allows us to track how far the sample has travelled, in addition, it also allowed it to sink into the gel.
- Set the voltage on the power supply (1-5V/cm between electrodes) to the required level. To cover the surface of the gel, add adequate running buffer. It’s crucial to utilize the same running buffer that you did for the gel preparation. When loading experimental materials, a suitable DNA size marker should always be included.
- Switch on the power supply and double-check that the gel box and the power supply are both operational. The anode should be closer to the wells than the cathode (black leads) (red leads). Activate the electrical system. Continue to run the gel until the dye has migrated to appropriate area.
Visualization
Turn off the power and remove the lid of the gel box when the electrophoresis is completed. Get the gel out of the gel box. Remove any excess buffer from the gel’s surface. To absorb any surplus flowing buffer, place the gel tray on paper towels. Remove the gel from the gel tray and place it in front of a UV light source. The most typical method is to use a gel documentation system. Orange luminous bands should appear as DNA bands. Dispose of the gel and running buffer in accordance with your institution’s policies.

Fig: Band of DNA under UV transilluminator

Fig: The illustration above depicts a typical result of DNA electrophoresis. On the left, there is a size marker that is used as a reference for the length of the sample DNA fragments (in base pairs). To the right of the marker are three samples: Sample A, Sample B and Sample C. The image shows how smaller DNA fragments move further through the agarose gel than the larger fragments of DNA. B) The graph to the right of the image shows the nonlinear, relationship between the size of the DNA fragments and the distance migrated. It is a negative curve, and as DNA fragments get larger, they migrate less distance through the gel.
Application:
- Agarose gel electrophoresis is used to resolve DNA, RNA, and protein fragments on the basis of their molecular weight.
- It is widely used for separation of DNA and RNA samples in events like restriction fragment analysis, polymerase chain reaction product analysis, checking the integrity of genomic DNA, molecular genetic diagnosis or genetic fingerprinting, and purification of nucleic acids.
- Separation of restricted genomic DNA prior to Southern blotting, or of RNA prior to Northern blotting.
- Protein separation, such as in clinical chemistry for the detection of protein abnormalities.
- One of the prominent applications of electrophoresis is separating DNA fragments for DNA sequencing and Phylogeny analysis.
- Even multiple topological forms of DNA, such as supercoiled, circular, or naked DNA, can be identified, separated, and characterized using the current technique.
- Besides, the agarose gel electrophoresis applies in other techniques, methods and fields are, distinguish homozygous and heterozygous, to find out the allele or allelic variations, and marker assisted selection- RFLP, AFLP and RAPD
References:
- Stellwagen, N. C. (2009). Electrophoresis of DNA in agarose gels, polyacrylamide gels and in free solution. Electrophoresis, 30(S1). Doi:10.1002/elps.200900052.
- Lee, Pei Yun et al. “Agarose gel electrophoresis for the separation of DNA fragments.” Journal of visualized experiments: JoVE ,62 3923. 20 Apr. 2012, doi:10.3791/3923
- Smith, S., Aldridge, P., and Callis, J. (1989). Observation of individual DNA molecules undergoing gel electrophoresis. Science, 243(4888), 203-206. Doi:10.1126/science.2911733.
- Stellwagen, N. C. (2009). Electrophoresis of DNA in agarose gels, polyacrylamide gels and in free solution. Electrophoresis, 30(S1). Doi:10.1002/elps.200900052.
- Rio DC, Ares M, Hannon GJ, Nilsen TW. Nondenaturing agarose gel electrophoresis of RNA. Cold Spring Harb Protoc. 2010;2010(6):pdb. prot5445. doi:10.1101/pdb.prot5445