
Introduction:
The accumulation of ungraded molecules as a result of a deficiency in a particular enzyme characterizes the rare inherited metabolic illnesses known as storage diseases. They are classified based on the site of aggregation, the type of substrate, and their clinical manifestations. MPS, GM1-gangliosidosis, mucolipidosis (ML), oligosaccharidosis, Pompe disease, Gaucher disease, Fabry disease, Niemann-Pick diseases, and neuronal ceroid lipofuscins are examples of lysosomal storage diseases
Niemann-Pick diseases (NPD) are a group of fatal inherited metabolic diseases of childhood, first described by Albert Niemann and further characterized by Ludwig Pick.
Niemann-Pick disorders, a sphingolipid disorder, are characterized by the build-up of toxic amounts of fatty substances called lipids, specifically sphingolipids and cholesterol, in various organs including the brain.
Normally, specific enzymes in the body break down lipids, whereas people with Niemann-Pick disease either lack these enzymes or the enzymes do not function properly, causing lipids to accumulate within cells. As the lipids build up in the cell compartment called the lysosome, Niemann-Pick diseases belong to a larger group of diseases called lysosomal storage disorders.
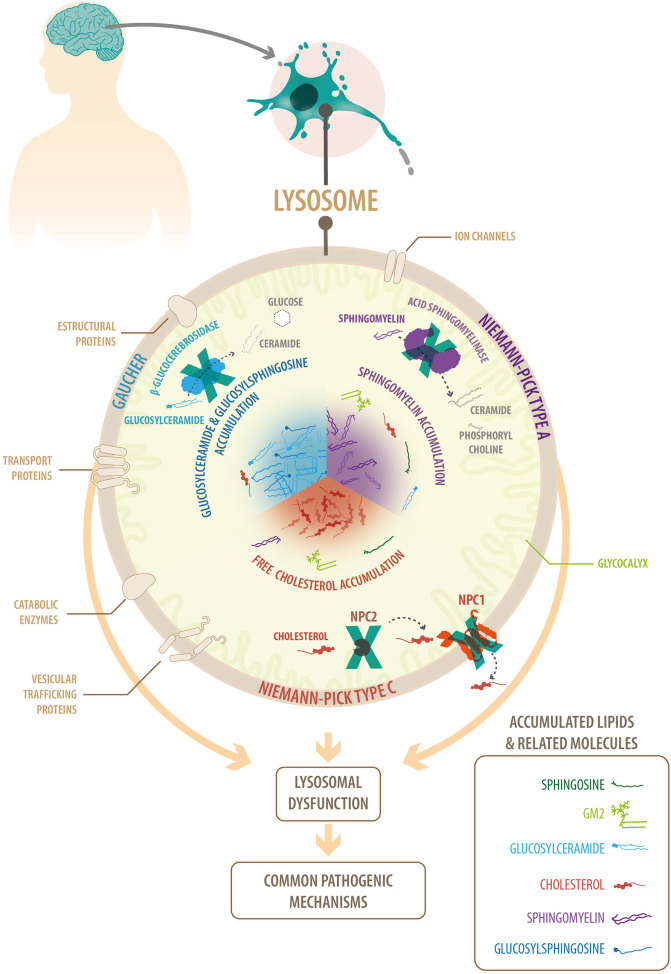
Fig: Common features of lysosomal neurodegenerative diseases: Niemann-Pick A, Niemann-Pick C
Genetic:
- It is caused due to recessive mutations in the SMPD1 gene on chromosome 11p15.4.
- These diseases are passed on by an autosomal recessive mode of inheritance, which means that when both parents carry a copy of the disease-associated gene, there is a 25% chance that their child will be affected.
- NPD is due to recessive mutations in the SMPD1 gene on chromosome 11p15.4Mutations in SMPD1 cause a lack of acid sphingomyelinase, resulting in less sphingomyelin breakdown and accumulation of this fat in cells. This fat accumulation causes cells to malfunction and finally die. In persons with Niemann-Pick disease types A and B, cell loss gradually compromises the function of tissues and organs such as the brain, lungs, spleen, and liver.
- Therefore, genetic testing is recommended for prospective parents who may be at a higher risk of being carriers of the disease-associated genes. For instance, the incidence of Niemann-Pick Type A is approximately 1 in 40,000 in Ashkenazi Jews, whereas Type D more commonly occurs in French Canadian populations from Nova Scotia. The overall incidence of Niemann-Pick disease is estimated to be approximately 1:100,000.
Type:
Niemann-Pick diseases are sub-classified into the types A, B, C and D, which differ by the genes in which the disease mutations occur, the type and severity of symptoms, as well as the age of onset.
Niemann Pick Disease Type A: classic infantile
Type A, which occurs in early infancy, is the most severe form of the disease. Symptoms include enlargement of the liver, spleen and lymph nodes, as well as significant damage to the brain.
Niemann Pick Disease Type B: visceral
Type B usually begins in childhood, with milder symptoms and no brain damage. Types A and B are caused by mutations in a gene called SMPD1, which results in ineffective activity of the enzyme that breaks down the lipid sphingomyelin.
Niemann Pick Disease Type C: subacute / juvenile
Niemann Pick Disease Type D: Nova Scotian
They are caused by a mutation in genes called NPC1 and NPC2, resulting in defective cholesterol transport in the brain. Types C and D may develop early or later in life, and are characterized by symptoms including learning difficulties, seizures, unsteady gait, slurred speech, difficulties with swallowing and eye movement, tremors and muscle loss, as well as liver and spleen enlargement.
Other Names for This Condition:
- Lipid histiocytosis
- Neuronal cholesterol lipidosis
- Neuronal lipidosis
- NPD
- Sphingomyelin lipidosis
- Sphingomyelin/cholesterol lipidosis
- Sphingomyelinase deficiency
Diagnosis:
Diagnosis can be performed by biochemical or microscopic tests in people presenting with symptoms of Niemann-Pick disease. Blood and bone marrow samples can be used to measure the amount of the enzyme ASM, which is defective in patients with Niemann-Pick disease Types A and B. Types C and D are diagnosed by examination of the cholesterol storage capabilities of skin cells grown from patients. Genetic testing may also be performed to determine whether mutations exist in the genes that are defective in Niemann-Pick disease. Prenatal testing is also available in some centres.
Treatment:
- There are limited available treatments for Niemann–Pick disease, and supportive care through nutrition and physical therapies can improve the quality of life for patients. Additionally, for patients with peripheral organ pathology, specialists can assist in reducing the severity of symptoms. Misglustat, registered as Zavesca, is an inhibitor of sphingolipid production that has recently been approved for treatment of Niemann-Pick C in Canada, the European Union, South Korea, Brazil, Russia and Australia. This drug does not cure the disease, but has been shown to slow neurological symptoms and disease progression in some patients.
- The National Institutes of Health (NIH) and Therapies for Rare and Neglected Diseases (TRND) are in the process of initiating human clinical trials to test the efficacy of cyclodextrin as a treatment for Niemann-Pick type C.
- Cyclodextrin has shown immense promise in correcting cholesterol imbalances, reducing neurodegeneration and prolonging life in mouse models of Niemann-Pick C disease, and has been trialled in two patients in the United States.
- Recently, experimental therapies including stem cell transplantation have been trialled in individuals with Type A and B disease, with varying degrees of success. While transplants have been successful at alleviating disease symptoms in two patients with Type B disease, complications due to the graft itself commonly arise as a consequence of treatment.
- Recent developments in the treatment of inborn metabolic defects, such as enzyme replacement therapy, substrate deprivation, pharmacological chaperone therapy, stem cell transplantation, and gene therapy, have increased optimism for the development of future remedies.
Prognosis:
Niemann-Pick Type A is the most severe type of the disease, with an average life expectancy of 18 months. Type B is a milder type of the disease, and some patients live into their late to mid-teens, with a few surviving into adulthood. Prognosis for patients with Type C varies between individuals and may range between late childhood through the mid to late teens, with some patients living into adulthood.
References:
- Aghamahdi, F., Nirouei, M. and Savad, S., 2022. Niemann-Pick type A disease with new mutation: a case report. Journal of Medical Case Reports, 16(1), pp.1-4.
- El-Gharbawy, A. and Vockley, J., 2017. Nonmitochondrial Metabolic Cardioskeletal Myopathies. In Cardioskeletal Myopathies in Children and Young Adults (pp. 265-303). Academic Press.
- Rimoin, D.L., Pyeritz, R.E. and Korf, B. eds., 2013. Emery and Rimoin’s essential medical genetics. Elsevier.
- Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. Int J Clin Pharmacol Ther. 2009;47.
- Schuchman, E.H. and Desnick, R.J., 2017. Types a and B Niemann-pick disease. Molecular genetics and metabolism, 120(1-2), pp.27-33.
- Levran O, Desnick RJ, Schuchman EH. Niemann-Pick disease: a frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patients. Proc Natl Acad Sci U S A. 1991 May 1;88(9):3748-52.
- Yañez, M.J., Marín, T., Balboa, E., Klein, A.D., Alvarez, A.R. and Zanlungo, S., 2020. Finding pathogenic commonalities between Niemann-Pick type C and other lysosomal storage disorders: Opportunities for shared therapeutic interventions. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1866(10), p.165875.