Introduction:
Some of the most powerful approaches to determine the taxonomy and phylogeny of the organisms is through the study of nucleic acids and proteins, since they are genes themselves or direct gene products that provide considerable information about the true relatedness.
Phylogenetic inferences based on molecular tools characterization provide the strongest and most reliable analysis of microbial evolution.
Nucleic acid base composition:
DNA base composition is widely employed for molecular based classification. DNA has four bases as Adenine, Guanine (purines) and Cytosine, thymine (pyrimidines) where Adenine pairs with Thymine by double hydrogen bonds and Cytosine pairs with Guanine by triple hydrogen bonds.
In DNA, G+C content is employed for characterization of the organism, based on the melting temperature of DNA that indicates the stability of DNA. This is because Guanine pairs with Cytosine with three hydrogen bonds so greater temperature is required to break open the double stranded DNA helix indicating high melting temperature(Tm) and stability.
Molecular % G+C = (G+C/ A+T+G+C) X 100%
G+C content is often determined by melting temperature of DNA. DNA melting can be followed spectrophotometrically to determine the absorbance of DNA under UV light in the wavelength of 260nm as DNA has highest absorbance in this wavelength.
As the single-strandedness of the DNA increases by heating, absorbance increases and steady state is gained when all the DNA length has become single stranded. This is hyperchromicity of DNA.
- Absorbance:ds DNA < ss DNA
- G+C content can also be determined by DNA hydrolysis and base analysis using HPLC.
- G+C content of DNA of animals and higher plants average around 40% and range between 30-50%. DNA of both eukaryotes and prokaryotes varies greatly in G+C content.
- Prokaryotic G+C content mostly ranges from 25-80%. Example: Actinomycetes: 59-73%, Bacillus: 32-62%, Treponema: 25-53%
- If two organisms differ in G+C content by more than 10%, their genome have quite different base sequences.
Significance
- It can only confirm the taxonomic data generated by other tools.
- Useful in generic classification only, as strains within the same species have constant G+C content.
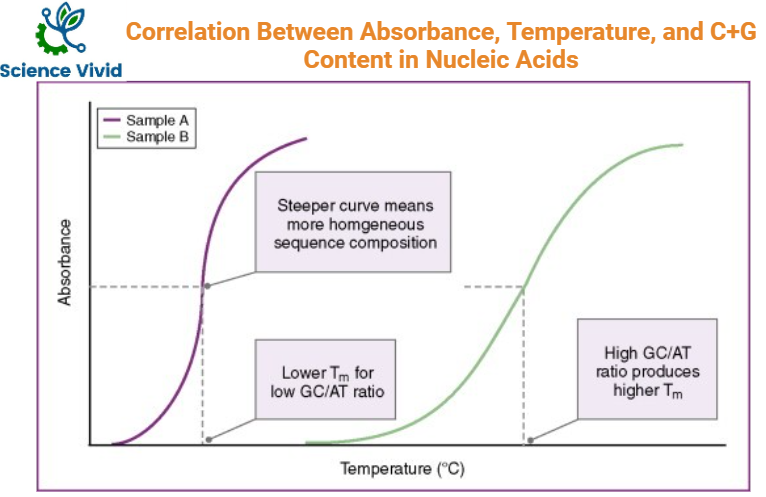
Fig: A graph showing Absorbance, Temperature, and C+G content correlation
Nucleic acid sequencing:
It simply means a determination of the sequence of nucleotides in DNA or RNA. Nucleic acid sequences are unique for each kind of organism. The smaller ribosomal subunit 16S rRNA from prokaryotes and 18S rRNA from eukaryotes are employed for determining the microbial taxonomy and phylogeny as they play a role in all organisms, and the ribosome is the symbol of the viability of a cell.
Certain signature sequences in Small subunit ribosomal ribonucleic acid (SSU rRNA) among organisms and other regions are quite stable due to slow change in gene, enabling the variable regions to be compared in distantly related organisms. Culturing the cells is not necessary. One can use environmental samples.
Strains with 16S rRNA sequence similarities <97% belong to different species, though sequence similarity >97% does not necessarily ensure the same species.
It can simply be performed using PCR and comparing its relatedness with different species. PCR amplifies the gene encoding 16S rRNA using rRNA primers, followed by primer extension with DNA polymerase, and sequenced using ddNTPs labelled with fluorescent agents. Strands of different lengths are formed during PCR. Then, Gel Electrophoresis is used to separate strands by size, and computer analysis all fluorescent signals. Then the sequences are compared with preexisting sequence data in RDP. Sab Value is calculated to compare relatedness. The higher the Sab value, the more closely the organism is related.
Significances
- Used to construct molecular phylogenies and classification at the genus and family level.
- DNA sequences can be obtained for single-copy genes, mitochondrial DNA, Ribosomal DNA, etc
Amino acid sequencing:
Amino acid sequences in proteins are a direct reflection of mRNA sequences as incoming amino acids are added by reading the codons in mRNA by recognition with anticodons of tRNA. So the most direct approach to compare proteins is to detect the amino acid sequences of proteins with the same function and the sequences of proteins with different functions. If the sequences of proteins with the same functions are similar, the organisms possessing them may be of similar types.
In 1966. It was demonstrated that protein-coding genes are often polymorphic, and gel electrophoresis of proteins could provide information on genetic variability (enzymes) in humans.
Inmmunological technology provides qualitative and quantitative estimate of amino acid sequences, differences in homologous proteins.
Advantages
- For the same homologous region, the amino acid sequence provides information about the elements that are more susceptible to micro/macro environments.
- Due to genetic code and Wobble hypothesis, most third-position mutation do not change the resulting amino acid. So any two amino acids sequences will be more conserved and similar than nucleotide sequences.
- The sequence of 20 amino acids has more information per site than a sequence of four nucleotides.
- Protein sequences are less affected by organism-specific differences in G+C content than DNA and RNA sequences.
- Important in species, subspecies, and strain level classification.
Nucleic acid hybridization:
Nucleic acid hybridization includes DNA-DNA homology or DNA base sequence and RNA sequence.
DNA-DNA hybridization (DDH) techniques have been used by taxonomists since the 1960s for the classification of prokaryotes. It has been considered as a gold standard method for determining the extent of relatedness between a set of strains, through hybridization of their respective genomic sequences and evaluation of resulting hybrids for degree of association or thermal stability. The recommended cutoff point for DDH similarity to define a new species is less than or equal to 70%.
If the mixture of single stranded DNA is cooled and held at temperature 25 degrees below the Tm, strands with complementary base sequences will reassociate to form stable dsDNA and under incubation at 10-15 degrees below Tm, hybrid formation occurs.
There is no fixed rule to determine if the two organisms fall in the same taxonomic rank if a certain percentage of hybridization is gained.
Under ideal hybridization conditions, two strains are regarded as belonging to the same species if they exhibit at least 70% relatedness and have a Tm difference of less than 5%.
Significances
- Used in genus, species, and subspecies level classification.
- Inability to produce cumulative databases
- Often criticized as cumbersome and inaccurate.
- Superior method for establishing bacterial species.
- Less costly than procedures like sequencing.
Genomic fingerprinting:
The DNA technique is used for comparing the nucleotide sequences of fragments of DNA
from different sources. The fragments are obtained by treating the DNA with various methods.
Endonucleases, enzymes that break DNA strands at specific sites, form patterns when subjected to gel electrophoresis. These patterns are called fingerprints.
Restriction fragment length polymorphism (RFLP), AmpFLP (Amplified fragment length polymorphism), and Short Tandem Repeats (STR) are some fingerprinting techniques.
By virtually hybridizing 191 fully sequenced bacterial genomes with a set of 15,264 13-mer probes specifically intended to create universal whole genome fingerprints, in silico genomic fingerprints were created. On the basis of comparing genomic fingerprints, a unique method for building phylogenetic trees was created. The bacterial phylogenetic tree that was generated was strikingly comparable to those that were generated by aligning conserved sequences using the Clusters of Genes of Corynebacterium and Bacillus.
Prokaryotic 16S rRNA fingerprinting can be achieved via oligodeoxyribonucleotide microarray and virtual hybridization, which can result in hybrid fingerprinting for identification.
With RFLP analysis, clonal populations, heterozygosity, relatedness, hybridization, and phylogenies with ages ranging from 0 to 50 million years ago can all be economically analyzed.
Steps in Genomic fingerprinting:
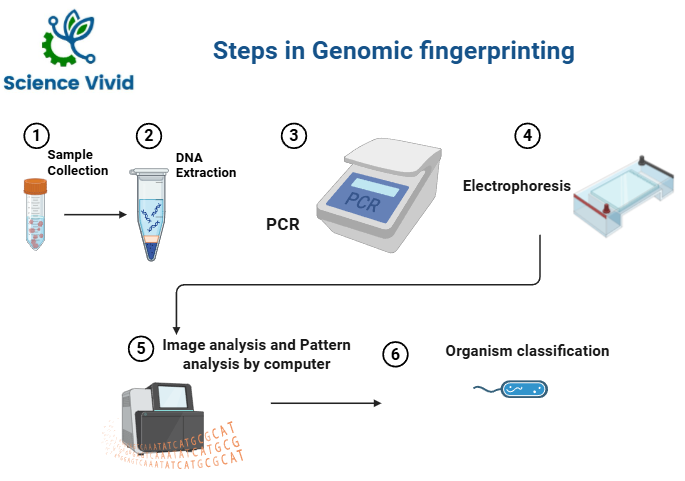
Fig: Steps in Genomic fingerprinting
Bacterial classification is mainly built on phenotypic features. When many of this data are compared by numerical taxonomy, a fair picture of relatedness can be seen. However, it is nearly impossible to project this picture back into past as numerical taxonomy covers only 20% of bacterial genome and orthodox bacterial taxonomy even less. So molecular methods with phylogenetic study provides best reliance in taxonomy and phylogeny.
References
- Blanco, A. & Blanco. (2022). Medical Biochemistry (2nd ed).
- Jaimes-Díaz, H., García-Chéquer, A. J., Méndez-Tenorio, A., Santiago-Hernández, J. C., Maldonado-Rodríguez, R., & Beattie, K. L. (2011). Bacterial classification using genomic fingerprints obtained by virtual hybridization. Journal of microbiological methods, 87(3), 286–294. https://doi.org/10.1016/j.mimet.2011.08.014
- National Institutes of Health. (2025, May 19). Ribosome. https://www.genome.gov/genetics-glossary/Ribosome
- National Institutes of Health. (2025, May 19). DNA sequencing. https://www.genome.gov/genetics-glossary/Ribosome