Mechanism of induction of cancer:
It is a gradual multistep process involving many generations of cells.
The cancer cells are genetically and phenotypically transformed cells having phenotypic features of malignancy like excessive growth, invasiveness, and distant metastasis. Normal cell growth is genetically controlled by four different types of regulatory control genes.
- Growth promoting oncogenes
- Growth inhibiting genes or tumor suppressor genes (anti-oncogene genes)
- DNA repair genes
- Genes regulating apoptosis
The abnormality in these four genes result in carcinogenesis:
- Activation of growth promoting oncogenes
- Inactivation of tumor suppressor genes
- Defective DNA repair genes
- Alteration in genes regulating apoptosis
Mechanism of Carcinogenesis:
Activation of growth promoting oncogenes. Proto Oncogenes are the normal genes which stimulate cell division.
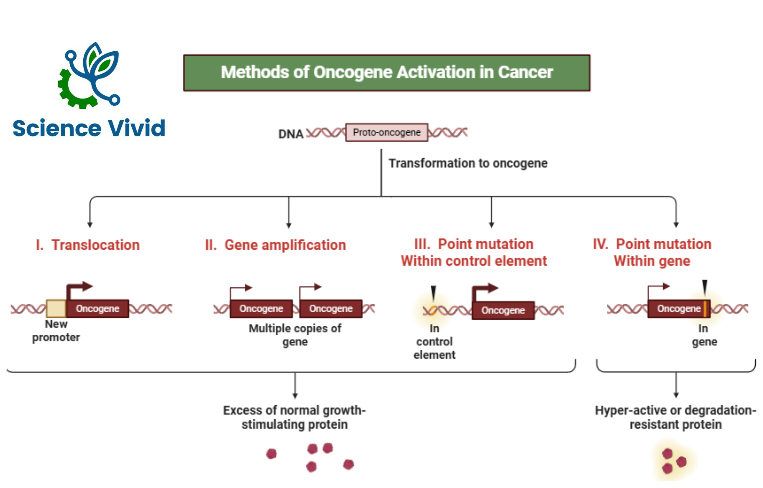
Cellular proto-oncogenes code for the number of proteins (growth factors, receptors, transcription factors and other proteins that are involved in cell proliferation. These protooncogenes are responsible for normal growth and development. But, when these protooncogenes get mutated by several mechanisms by point mutation, deletion, duplication, chromosomal translocation, gene amplification, they become oncogene.
And these oncogenes are the gene which are capable of causing cancer and when these oncogenes are expressed, they result in the formation of oncoproteins and there is formation of mutated versions of all growth factors, and receptors and it ultimately result in malignant growth of cells. Thus, oncogene can be defined as an altered gene, the product of which acts in a dominant manner to accelerate cell growth or cell division.
Proto Oncogenes are activated in oncogene by several mechanisms like
- Promoter insertion
- Chromosomal translocation
- Enhancer insertion
- Gene amplification
- Point mutation
Promoter insertion
This is a schematic representation of the host genome having c-MYC gene. It is in an inactive state and it can lead to transcription of MYC-mRNA. But when retrovirus infect host cells the single stranded RNA of retrovirus is transcribed into single stranded DNA by the action of enzymes known as reverse transcriptase. Again, the single stranded DNA leads to formation of complementary DNA and this double stranded DNA is now called as pro-virus.
This viral genome now has long terminal repeat sequences which can act either promoter or enhancer. A promoter is a sequence of genes which is required to turn on or off the specific gene. The process of transcription is initiated at the promoter and they are usually found near the beginning of the gene. So, when the DNA genome or viral genome integrates into the host genome the enhancer region activates C-MYC. And there will be vigorous transcription of MYC-mRNA. This is how the promoter insertion leads to conversion of protooncogenes into oncogenes.
Likewise, in the enhancer insertion mechanism, the enhancer sequences are regulatory DNA sequences which enhance the transcription of associated genes. In this also, the viral genome is integrated into the host genome and acts as an enhancer and leads to vigorous transcription of MYC-RNA.
This is how both promoter insertion and enhancer insertion lead to activation of protooncogenes into oncogenes
Chromosomal Translocation
Chromosomal translocation occurs in Burkitt’s lymphoma. It is a cancer of B lymphocytes, two chromosomes are involved, there is translocation between chromosome 8 and chromosome 14.
Myc Gene is located in the long arm of chromosome 8 and igG H gene is located in the long arm of chromosome 14. So, the terminal segment of the long arm of chromosome 8 breaks off and it translocates to chromosome 14. The reverse process moves a small segment of chromosome 14 to chromosome 8.
So, the myc gene which was earlier in chromosome 8 is now translocated to chromosome 14 and it is placed adjacent to the gene transcribing the heavy chain of immunoglobulin molecules. So, this myc gene is now activated by this potent immunoglobulin H gene enhancer and this is how chromosomal translocation leads to activation of protooncogene into oncogene.
Chromosomal translocation also occurs in chronic myeloid leukemia (CML) which involves two chromosomes, chromosome 9 and 22. ABL gene is present in chromosome 9 and BCR gene in chromosome 22. The terminal segment of the long arm of chromosome 9 splits off and it translocates to chromosome 22 and reverse occurs, the segment from chromosome 22 splits and then joins to chromosome 9. And this chromosomal translocation now leads to change in both the chromosomes, so this chromosome 22 is having both BCR and ABL and this chromosome is now known as the Philadelphia chromosome.
Gene amplification
Normally the copies of genes are transcribed and translated to form normal proteins and there is controlled cell growth.
But, in cases of gene amplification, many more copies of genes are formed than normal and it ultimately results in increased protein activity and uncontrolled cell growth. So, this is how gene amplification also plays a major role in activation of protooncogene to oncogene.
Point mutation
Mutation is change in the nucleotide sequence of DNA and point mutation is an important mechanism of activation of protooncogene into oncogene.
The classical example of point mutation is RAS oncogene.
Mechanism of action of oncogene:
When a growth factor interacts with a growth factor receptor it leads to activation of binding protein, it can be a tyrosine kinase enzyme and inactive RAS which is associated with the cell membrane becomes activated and this activated RAS protein activate the RAF/MAP kinase pathway of PI3 kinase pathway and leads to transcription of genes.
Tumor suppressor Genes:
Tumor suppressor genes code for protein which function to control cell division and ensure that it only occurs when necessary. It is also recognized as a guardian of gene.
Tumor suppressor genes code for proteins involved in preventing cell cycle progression known as gate keepers and proteins involved in repairing DNA known as care takers.
Normally, these proteins prevent accumulation of DNA mutations by repairing DNA mutations when they occur and preventing cells with DNA mutations from replication. When mutations inactivate the function of these proteins, DNA mutations can accumulate. This leads to cancer.
Mutations which lead to loss of function of tumor suppressor genes increase the risk of cancer by increasing the risk of uncontrolled cell division.
p53 gene
- P53 is the most important suppressor gene, in conjugation with other protein ensures that only cells free of DNA errors can undergo cell division.
- If a cell contains DNA errors, p53 will either arrest cell division until the DNA has been repaired or it will trigger apoptosis of that cell.
- Loss of p53 leads to accumulation of DNA mutations and eventually.
- p53 gene: The Guardian of the genome
- p= protein, 53 weight of the protein 53kDa discovered in 1978.
- It is reported that there is almost 50% pf p53 mutation in all human tumors.
Function
P53 is a tumor suppressor protein that play a key role in
- Regulation of the cell ycle or growth by applying brakes to the cell proliferation
- Apoptosis (programmed cell death)
- Maintenance of genome stability
- Recruits and trigger DNA repair gene
- Loss of the function of these genes is a key event in carcinogenesis.
- Prevent neoplastic transformation either by cell cycle arrest or by triggering apoptosis
- It is activated in response to DNA damage and pauses the cell cycle.
Let’s say if there is a DNA damage that happens during G1 phase and if these particular damages aren’t repaired these could lead to a negative consequence because these are really deleterious in terms of nature.
Basically, it blocks replication fork, loss of chromosomal segment and worst outcome will be apoptosis and death of the cell. Thus, p53 can modulate the cyclins and prevent and cause pause in the cell cycle.
Basically, there are special sensors known as ATM (Ataxia telangiectasia mutated) and ATR (Ataxia telangiectasia and Rad3-related) which can sense and bind to DNA damage. With downstream signaling pathways like chk2 (check-2), p53 can be activated. Normally, p53 is degraded but when check-2 phosphorylates p53, it is activating and activated P53 can do numerous activities. It can activate p21, a negative influencer of cyclin/CDKs. So, when P21 blocks cyclin/CDKs activities the cell cycles halt and can’t progress further.
Pausing the cell cycle is very crucial as it could give DNA repair mechanisms enough time to repair damage. There is a homologous recombination mechanism and non-homologous end joining based mechanism by which DNA can be repaired. After, the damage is repaired, cell cycle is resumed and thereby cell cycle progression happens and cell divides accordingly.
However, if the DNA damage can’t be repaired in this case the cell will be induced for apoptosis. P53 coordinated with caspases-3 will basically target the intrinsic pathway of apoptosis. It activates Bax, Bad which poke holes in the mitochondrial membrane, cytochrome-c will leak and ultimately caspase-3 is activated which triggers the responses.
Is localized in the nucleus, its half-life is 20minutes. Gene is responsible for suppressor of tumors, i.e. It acts against the formation of tumors.
It encodes protein, regulates and suppresses cellular proliferation mainly by inhibiting progression of cell through cell cycle.
It is present in chromosome 17 and prevents cells with DNA damage from entering into S- phase, hence, preventing multiplication of mutated cells.
If there is a DNA damage, p53 recruit retinoblastoma (Rb) gene to stop the cell cycle to allow for repair. If the damaged DNA is not repaired p53 tells the cell to undergo apoptosis. Thus, p53 is called the “guardian of genome and molecular policeman” because it will never allow the cell to undergo abnormal proliferation. Because it plays a major role in
- Antiproliferative effect
- Apoptosis
- DNA repair
Mutation in the p53 gene causes the resistance in the tumor, mainly, to the radiation and chemotherapy.50 50% of human tumors contain mutations in p53 gene, where the most common mutation is in the DNA binding domain.
It is down-regulated by mdm2, where this particularly acts as an oncogene.
Another important protein called E6 of HPV that inactivates p53. Like, P53 there are also several gene such as p63 and p 73 which are also the family of p53 gene. In this p63 is responsible for differentiation of stratified squamous cell epithelia. p73 is the elder brother of p53 that is located at chromosome 1 and plays a major role in cell cycle and apoptosis which illustrate the similar effect like p53.
Rb gene
Retinoblastoma
First tumor suppressor gene to be discovered and the most commonly studied and is expressed in every cell type.
Mutation of the Rb gene occurs in the Rb pocket. Viruses such as polyoma, HPV, and adenovirus mainly bind to the RB pocket.
The loss of Rb gene is associated with retinoblastoma as well as osteosarcoma and the sporadic is 60% of cases and 40% of cases is reported to familial or hereditary.
Adenomatous polyposis Coli (APC) gene
It is recognized as the gatekeeper of colonic neoplasia, a critical tumor suppressor gene in the colon, especially down regulation of the WNT signaling pathway. This pathway is responsible for regulating several biological processes including cell pluripotency, tissue homeostasis and determines the differentiation fate of cells during development. Mutation in this gene is associated with familial adenomatous polyposis (FAP) and colorectal cancer. It is an autosomal dominant condition associated with deletion of APC gene which is located on chromosome 5q21.
Oncogenes:
- Oncogenes code for proteins that acts as powerful inducers of cell division and prevention of cell death (cell survival).
- Mutations which lead to gain of function of oncogenes increases the risk of cancer by promoting constant cell divisions and/or survival.
- Oncogenes which have not been mutated and are functioning normally are called proto-oncogenes.
- Oncogenes code for protein which are involved in signaling for cell division and cell survival such as growth factors, growth factors receptors, signal transduction proteins, and transcription factors.
- Normally, these proteins are switched off until cell division/survival is required. However, mutations can lead to situations in which they are permanently turned on.
Inherited cancer syndromes:
- Approximately 5-10% of all cancer instances are familial or inherited.
- Approximately 90-95% of all cancer instances are sporadic.
- Most causes of inherited or familial cancer are due to germline mutation in tumor suppressor gene.
Somatic vs Germline mutations:
Mutations are categorized based on whether they occur in germline cells (sperm and eggs) or non-germline cells (every other cell type). Germline mutations occur in sperm and eggs cells and therefore can be inherited.
Somatic mutations occur in non- germline cells. The mutations are not inheritable; however, they are responsible for most cases of cancer (90% of all cancers).
Two hit Hypothesis:
- For a cell to progress towards cancer, both copies of tumor suppressor must be activated.
- In cases of inherited cancer syndromes, children inherit a germline mutation in one copy of their tumor suppressor gene. This is the first hit.
- Somatic mutation inactivates the remaining functional tumor suppressor gene. This is the second hit.