Introduction:
Alport syndrome is a rare hereditary illness characterized by increasing renal dysfunction, sensorineural hearing loss, and ocular abnormalities, and visual problems. In the early twentieth century, the condition now known as Alport syndrome was originally documented in the British medical literature. Dr. Cecil Alport released a paper in 1927 outlining the relationship between kidney disease and deafness in affected persons. Numerous more examples were recorded in the literature, and in 1961, the condition was named after Dr. Alport.
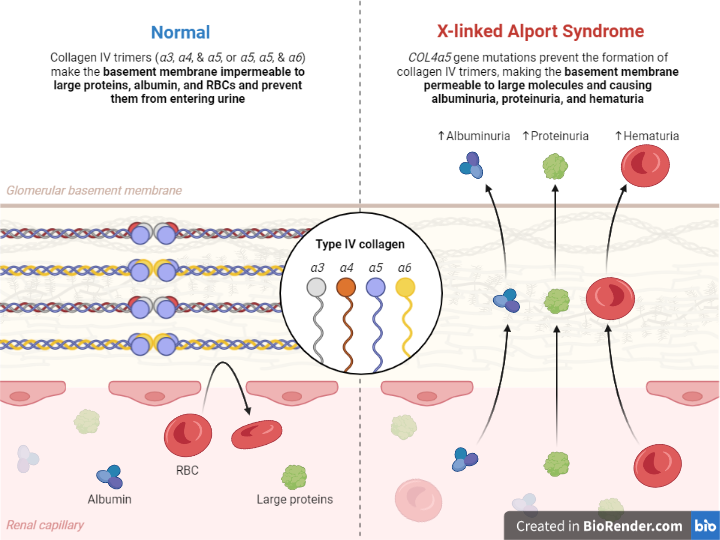
Fig: Alport syndrome
Incidence:
X-linked Alport syndrome (XLAS) is believed to affect 1 in 50,000 to 1 in 5,000 people. Although XLAS is considered an uncommon condition, it may be more prevalent in particular populations. For example, the prevalence of XLAS in African-American men can be as high as 1 in 2,500. XLAS is also more common in other European countries, such as the Basque Country, where the incidence might reach 1 in 10,000 people.
XLAS can occur sporadically, which means there is no family history of the disorder, although it is more typically inherited in an X-linked recessive pattern. Because of the greater likelihood of inheriting the mutant gene in families with a history of XLAS, the prevalence can be higher.
Cause:
X-linked Alport syndrome (XLAS) is caused by mutations in the COL4A5 gene on the X chromosome. The COL4A5 gene codes for a protein termed type IV collagen alpha-5 chain, which is a component of basement membranes in the kidneys, ears, and eyes. These basement membranes give structural assistance and help in the removal of waste materials from the bloodstream. Mutations in the COL4A5 gene result in a defective type IV collagen alpha-5 chain protein, which impairs the integrity and function of the basement membranes.
People with XLAS have progressive impairment to the filtering units (glomeruli) of their kidneys, resulting in proteinuria (protein in the urine), hematuria (blood in the urine), and, eventually, kidney failure. XLAS can also impact the ears, causing hearing loss, as well as the eyes, producing vision issues such as cataracts and retinal abnormalities.
XLAS is inherited as an X-linked recessive trait, which means that the defective gene lies on the X chromosome. Because males only have one X chromosome, they will have XLAS if they acquire a mutant COL4A5 gene from their mother.
Females have two X chromosomes; therefore, they can inherit a mutated gene on one X chromosome and have a milder form of XLAS, or they can inherit mutant genes on both X chromosomes and have a more severe form of the condition.
Types:
There are several types of Alport syndrome, which are differentiated based on the mode of inheritance and the specific genetic mutation involved. The main types are:
X-linked Alport syndrome (XLAS): This is the most common form of Alport syndrome, accounting for around 80% of cases. It is caused by mutations in the COL4A5 gene, which is located on the X chromosome. As a result, XLAS primarily affects males, who have only one X chromosome. Females can also be affected, but the severity can vary widely depending on the degree of inactivation of the X chromosome carrying the mutated gene.
Autosomal recessive Alport syndrome (ARAS): This form of Alport syndrome is caused by mutations in either the COL4A3 or COL4A4 gene, which are located on chromosomes 2 and 13, respectively. ARAS is inherited in an autosomal recessive pattern, meaning that a person must inherit two copies of the mutated gene (one from each parent) to develop the disorder.
Autosomal dominant Alport syndrome (ADAS): This is the rarest form of Alport syndrome and is caused by mutations in the COL4A3 or COL4A4 gene, inherited in an autosomal dominant pattern, meaning that only one copy of the mutated gene is required to develop the disorder. ADAS is usually milder than other forms of Alport syndrome, and the onset of symptoms is typically later in life.
Non-classic Alport syndrome: This type of Alport syndrome is characterized by atypical clinical features and does not fit the classic diagnostic criteria for Alport syndrome. It can be caused by mutations in the same genes involved in classic Alport syndrome or other genes that affect the basement membrane.
Pathology:
Alport syndrome is a hereditary condition that mostly affects the basement membranes of the kidneys, ears, and eyes. Mutations in the genes encoding type IV collagen produce weaker and disturbed basement membranes in afflicted organs. This disruption causes hematuria, proteinuria, and kidney failure in the kidneys, hearing loss in the ears, and anterior lenticonus, cataracts, and retinal abnormalities in the eyes, which are all hallmarks of Alport syndrome.
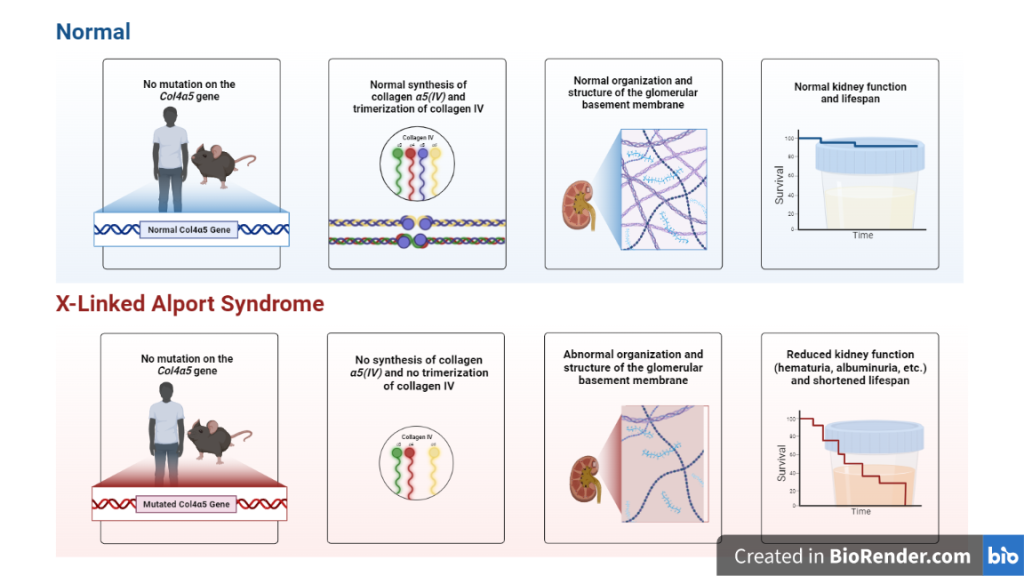
Fig: Pathology of Alport syndrome
The severity of Alport syndrome varies depending on the type of gene mutation and the degree of involvement of the affected organ. Alport syndrome is inherited in an autosomal recessive or X-linked recessive form, and there is currently no cure. Medication, hearing aids, or kidney transplantation may be used to treat symptoms and prevent problems.
Symptoms:
| Type of Alport Syndrome | Mode of Inheritance | Gene(s) Involved | Common Symptoms and Features |
| X-linked Alport syndrome (XLAS) | X-linked recessive | COL4A5 | Hematuria, proteinuria, progressive kidney dysfunction, sensorineural hearing loss, anterior lenticonus, cataracts, retinal abnormalities |
| Autosomal recessive Alport syndrome (ARAS) | Autosomal recessive | COL4A3 or COL4A4 | Hematuria, proteinuria, progressive kidney dysfunction, sensorineural hearing loss, eye abnormalities, end-stage kidney disease |
| Autosomal dominant Alport syndrome (ADAS) | Autosomal dominant | COL4A3 or COL4A4 | Hematuria, proteinuria, progressive kidney dysfunction, sensorineural hearing loss, eye abnormalities |
| Non-classic Alport syndrome | Variable | COL4A3, COL4A4, or other genes | Atypical clinical features, such as absence of hematuria or late onset of symptoms, eye abnormalities, hearing loss, and progressive kidney dysfunction |
Diagnosis:
Urinalysis: A urinalysis can detect the presence of protein and blood in the urine, which are common early signs of XLAS.
Kidney biopsy: A kidney biopsy involves taking a small sample of kidney tissue for examination under a microscope. This can help to confirm the presence of kidney damage, inflammation, and scarring, which are all common features of XLAS.
Audiometry: Audiometry is a hearing test that measures how well an individual can hear sounds of different frequencies and volumes. This can detect the progressive hearing loss that is a common feature of XLAS.
Eye exam: An eye exam can detect abnormalities such as thickening of the lens or detachment of the retina, which are common in individuals with XLAS.
Genetic testing: Genetic testing can identify mutations in the COL4A5 gene, which is responsible for XLAS. This can confirm a diagnosis and help to identify family members who may also be affected.
Family history: A family history of kidney disease, hearing loss, or eye abnormalities can suggest the presence of XLAS and may prompt further diagnostic testing.
Treatments:
Although there is no cure for Alport disease, existing therapies can alleviate some symptoms and delay kidney impairment. Patients should also follow a “renal healthy” diet that is low in salt, phosphates, and potassium. Dialysis or a kidney transplant are the only treatments available to patients with renal failure. Several of the eye defects that impede vision in XLAS sufferers can be treated surgically.
To monitor the progression of disease and treat symptoms, it’s highly recommended people with XLAS to follow up regularly with a nephrologist and other experts, such as an audiologist and an ophthalmologist. To assess the likelihood of passing the condition on to future generations, genetic counseling may also be beneficial.
References:
Zhang, X., Zhang, Y., Zhang, Y. et al. X-linked Alport syndrome: pathogenic variant features and further auditory genotype-phenotype correlations in males. Orphanet J Rare Dis 13, 229 (2018).
https://my.clevelandclinic.org/health/diseases/24202-alport-syndrome
https://myriad.com/womens-health/diseases/x-linked-alport-syndrome/