Background:
- Because ammonia is extremely poisonous, catabolic ammonia generation is a significant metabolic concern.
- Amino groups are directed into a single excretory end product if they are not reused again for the synthesis of new amino acids or other nitrogenous products.
- The majority of aquatic organisms, including bony fish, are ammoniotelic, which means they excrete amino nitrogen as ammonia.
- Animals living on land need nitrogen excretion systems that reduce toxicity and water loss. Birds and reptiles are uricotelic, excreting amino nitrogen as uric acid, while the majority of terrestrial animals are ureotelic, excreting amino nitrogen in the form of urea.
- The urea cycle in ureotelic organisms converts the ammonia accumulated in hepatocyte mitochondria to urea.
Introduction:
- The urea cycle is the primary route for nitrogen removal in humans.
- The end result of protein metabolism is urea (amino acid metabolism). The nitrogen in amino acids is toxic to the organism when transformed to ammonia. It is detoxified after being transformed to urea.
- As a result, urea constitutes 80-90 percent of the nitrogen-containing compounds excreted in urine. Urea is produced in the liver and transferred to the kidneys for excretion in the urine.
- The urea cycle was the first metabolic cycle discovered by Hans Krebs and Kurt Henseleit (1932), and it is also known as the Krebs-Henseleit cycle.
- Urea has two amino (NH2) groups, one from NH3 and one from aspartate. CO2 provides a carbon atom.
Occurrence:
In the mitochondria of liver cells, the urea cycle, also known as the ornithine cycle, turns excess ammonia into urea. Urea originates, enters the bloodstream, is filtered by the kidneys, and is eventually excreted in the urine.
Steps:
Urea production is a five-step cyclic process involving five different enzymes. The first two enzymes are present in the mitochondria of hepatocytes while the rest are localized in cytoplasm.
The several steps involved are as follows:
Synthesis of carbamoyl phosphate
The first step, which is also irreversible and rate-limiting, involves the conversion of CO2 and ammonia into carbamoyl phosphate via the enzyme carbamoyl phosphate synthetase I (CPS I).
Formation of citrulline
Carbamoyl phosphate and ornithine combine to form citrulline via ornithine transcarbamoylase (OTC). Ornithine translocase then transports citrulline from hepatocyte mitochondria into the cytoplasm.
Synthesis of arginosuccinate
Citrulline reacts with aspartate to form argininosuccinate. The ATP-dependent enzyme argininosuccinate synthetase is responsible for this process. The second amine group on urea is produced by aspartate.
Cleavage of arginosuccinate
Argininosuccinate is converted into arginine via argininosuccinate lyase. Fumarate is a byproduct of this reaction that is also used in the breakdown of tyrosine and the mitochondrial production of NADH for the TCA cycle.
Formation of urea
Arginase hydrolyzes arginine to produce urea and ornithine. Ornithine is thus regenerated and enters mitochondria for reuse in the urea cycle.
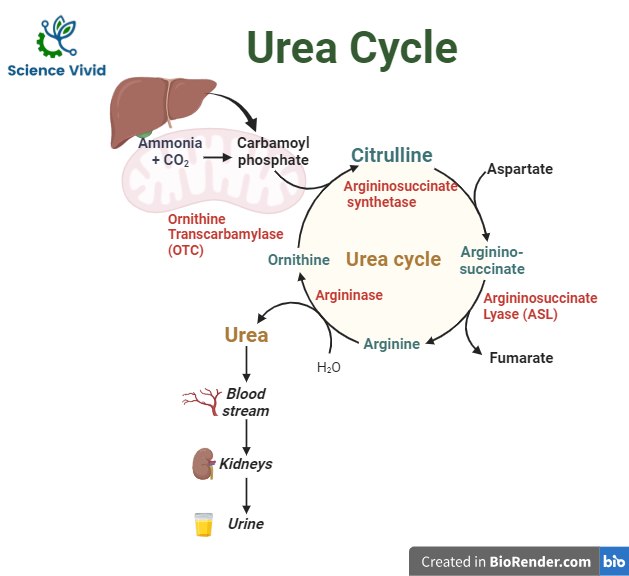
Fig: Urea cycle
Regulations:
- The urea cycle consists of five enzymatically controlled steps that are catalyzed by carbamyl phosphate synthetase, ornithine transcarbamylase, argininosuccinate synthetase, argininosuccinase, and arginase, respectively.
- The production of urea is primarily governed by two factors: the quantities of urea cycle enzymes and acetyl-glutamate and ornithine.
- The contents of all the urea cycle enzymes in the liver is directly proportional to the daily consumption of protein, then the activities of urea cycle enzymes are an important regulatory factor of the urea cycle.
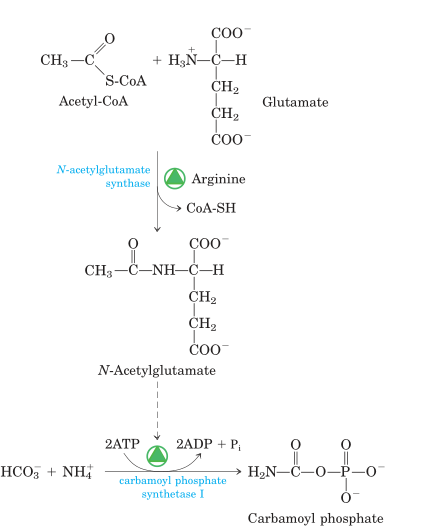
Fig: Synthesis of N-acetylglutamate and its activation of carbamoyl phosphate synthetase I.
- The first reaction catalyzed by carbamoyl phosphate synthase I (CPS I) is a rate-limiting reaction or committed step in urea production. N-acetylglutamate allosterically activates CPS I. (NAG). It is produced from glutamate and acetyl CoA by synthase and destroyed by a hydrolase. The rate of urea synthesis in the liver is related to the content of N-acetylglutamate. NAG is increased by high arginine concentrations. Consuming a protein-rich meal raises the level of NAG in the liver, resulting in increased urea production.
- Glucagon, insulin, and glucocorticoids are major regulators of the expression of urea cycle enzymes in liver.
Linkage of Urea cycle and TCA Cycle:
The citric acid cycle and the urea cycle are connected but separate cycles. The transamination of oxaloacetate to aspartate yields one of the nitrogen atoms used in the urea cycle. The fumarate that is produced in urea cycle is also an intermediate in the citric acid cycle.
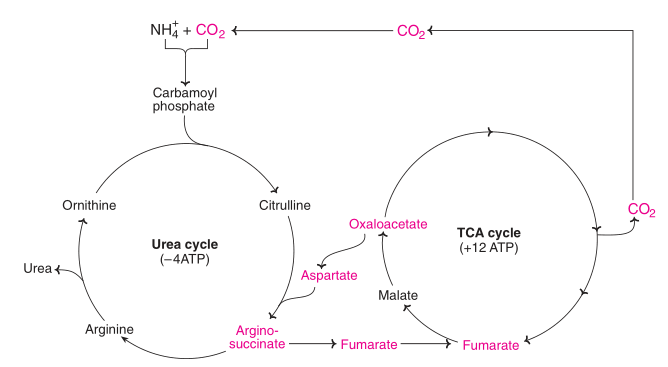
Fig: Interrelation between urea and tricarboxylic acid (TCA) cycle
Deficiencies:
- Symptoms in people with urea cycle abnormalities usually arise from the exposure of accumulated metabolites. The cumulative prevalence of these diseases is high (at least 1 in every 2000 infants). It is crucial to respond promptly and correctly during the initial presentation and subsequent occurrences.
- The urea cycle is made up of five enzymes that convert ammonia from amino acid metabolism to urea in a sequence of processes. Each of these enzymes has been observed to have genetic problems, and the overall incidence has been estimated to be 1 in 35,000 births, however partial deficiencies may raise the frequency significantly. Although Urea Cycle Defects (UCDs) syndromes are not associated with severe liver impairment, the core genetic abnormality is located inside the liver, and the symptoms can resemble those of other metabolic liver illnesses.
- Primary Urea Cycle Disorders (UCDs) include carbamoyl phosphate synthase (CPS) deficiency, ornithine transcarbamylase (OTC) deficiency, argininosuccinate synthetase deficiency (citrullinemia), argininosuccinate lyase deficiency (argininosuccinic aciduria), arginase deficiency The more the enzyme deficiency, the more severe the hyperammonemia; consequently, illness severity is NAGS deficiency, CPS deficiency, OTC deficiency, citrullinemia, argininosuccinic aciduria, and argininemia.
Clinical manifestations:
There are modest to severe clinical symptoms, such as intellectual impairment, failure to thrive, and episodic hyperammonemia (e.g., altered mental status, coma, death).
Growth failure, developmental delay, psychological problems, episodic (particularly postpartum) hyperammonemia, and phenotypes comparable to those of afflicted males are just a few of the symptoms that impact female carriers of ornithine transcarbamylase (OTC) deficiency may experience (i.e., recurrent vomiting, irritability, lethargy, hyperammonemic coma, cerebral edema, spasticity, intellectual disability, seizures, death).
- Slurred speech
- Disorientation
- Confusion
- Sleepy or sluggish
- Agitation
- Delirium
- Lethargy
- Stroke-like symptoms
- Psychiatric issues like
- bipolar
- disorder and
- schizophrenia
Treatment of Urea Cycle Disorders:
The therapy of urea cycle disorders is a lifetime process that aims to manage symptoms rather than cure the condition. Monitoring ammonia levels with blood tests – serum and plasma levels – at regular intervals is one strategy, and the data are used to optimize treatment procedures.
- Dietary protein restriction
- Arginine or citrulline supplementation
- Sodium phenylbutyrate
- Possible liver transplantation
References:
- Fearing, M.K. and Shih, V.E., 2004. Urea Cycle, Inborn Defects of.
- Satyanarayana, U., 2021. Biochemistry, 6e-E-book. Elsevier Health Sciences.
- David, L., Nelson, D.L., Cox, M.M., Stiedemann, L., McGlynn Jr, M.E. and Fay, M.R., 2000. Lehninger principles of biochemistry.
- Saheki, T., Hosoya, M., Fujinami, S. and Katsunuma, T., 1982. Regulation of urea synthesis: changes in the concentration of ornithine in the liver corresponding to changes in urea synthesis. In Urea Cycle Diseases (pp. 255-263). Springer, Boston, MA.
- Shambaugh III, G.E., 1977. Urea biosynthesis I. The urea cycle and relationships to the citric acid cycle. The American journal of clinical nutrition, 30(12), pp.2083-2087.