Introduction:
The use of metagenomics allows for direct access to the genetic material of whole communities of organisms using a variety of genomic technologies and bioinformatics tools. The direct genetic examination of genomes present in an environmental sample is referred to as metagenomics.
The study of whole nucleotide sequences that have been extracted and examined from all of the organisms (usually bacteria) in a bulk sample is known as metagenomics. It is frequently used to investigate a particular population of microbes, such as those found in soil, water, or on human skin.
The goal of metagenomics is to better understand the variety, structure, and function of microbial communities in many habitats, such as soil, water, and the human gut. Researchers can assess the genetic content of large communities of microbes, as well as find new and previously unknown species, metabolic pathways, and functions. Metagenomic analysis gives a thorough understanding of microbial diversity and function in environmental samples and can be applied to a variety of applications such as environmental monitoring, disease diagnostics, and biotechnology.
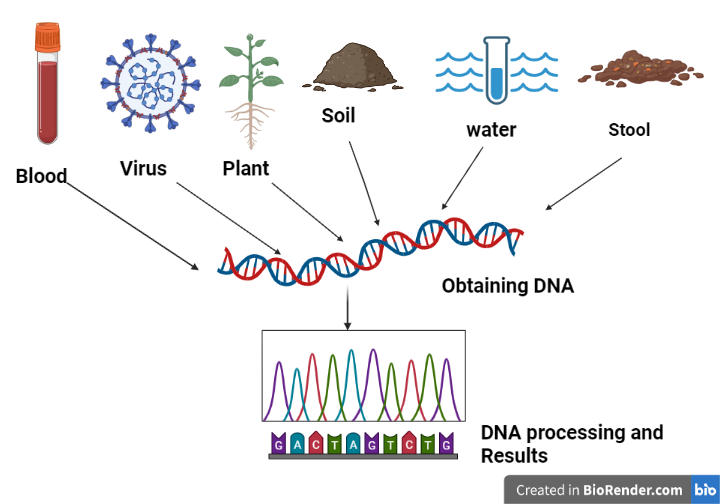
Fig: The process of metagenomic analysis
Methods for characterising metagenomics:
- High-throughput sequencing: This method involves the sequencing of DNA from the environmental sample, to obtain a comprehensive view of the microbial community.
- Taxonomic profiling: This method involves the classification of sequences based on their taxonomic origin, to understand the diversity and abundance of microorganisms present in the sample.
- Functional analysis: This method involves the annotation of genes and the prediction of their functions, to understand the metabolic capabilities of the microbial community.
- Network analysis: This method involves the construction of interaction networks between microorganisms, to understand the functional relationships between them.
- Metabolic modelling: This method involves the reconstruction of metabolic networks, to understand the metabolic interactions between microorganisms and their role in the environment.
- Comparative metagenomics: This method involves the comparison of metagenomic datasets from different samples or environments, to identify common and unique features of the microbial communities.
Steps:
Sample collection and DNA extraction
Sample collection is an important step in metagenomics research. A sample of the environment (known as eDNA) is obtained. A soil sample, a water sample, a urine sample, a faeces sample, and a gut sample are collected. Collect a sample from the environment and extract DNA from the collected cells. Physical separation and isolation of cells from samples may also be necessary to enhance DNA yield or minimize coextraction of enzyme is that could interfere with later processing.
Quality control and sequencing
Perform quality control checks on the extracted DNA. The extracted DNA should be representative of all cells in the sample, and adequate amounts of high-quality nucleic acids should be collected for subsequent library construction and sequencing. If the quantity of extracted DNA is insufficient in the samples (such as biopsies or ground-water). It is highly recommended to amplify the fragments using PCR and then construct a DNA library for further down streaming.
Sample analysis
PCR
PCR Amplification: Polymerase chain reaction (PCR) is often used to amplify specific regions of the DNA for further analysis. PCR can be used to amplify 16S rRNA genes, which are widely used for bacterial identification, or other genes that are specific to the organisms of interest.
DNA sequencing
High-Throughput Sequencing: High-throughput sequencing technologies, such as Illumina, PacBio, and Oxford Nanopore, are commonly used for metagenomic sequencing. These technologies allow for the simultaneous sequencing of millions of DNA fragments, enabling the identification of all the organisms present in a sample.
DNA microarray
DNA microarray technology can also be used for sample analysis in metagenomics. DNA microarrays are small chips that contain thousands of probes that can detect specific DNA sequences. DNA microarrays have several advantages for metagenomics sample analysis, including the ability to analyse a large number of samples simultaneously, the ability to detect multiple organisms in a single assay, and the potential for high-throughput analysis. However, they also have some limitations, such as a limited dynamic range and the requirement for prior knowledge of the DNA sequences of interest.
Sequence assembly
Sequence assembly is an important stage in metagenomics, which aims to reconstruct the genomes of microbial communities found in environmental samples. Because it involves processing and merging millions of short DNA reads from many distinct organisms with varied abundances, metagenomic sequence assembly is more difficult than standard genome assembly. Following the assembly of metagenomic sequences into contigs, many post-assembly procedures are carried out, including contig validation, scaffolding, and gap filling. The post-assembly processes are critical for increasing the metagenomic assembly’s accuracy and completeness. Overall, sequence assembly is an important stage in metagenomics, and the approach used is determined by the complexity and diversity of the microbial community, available computer resources, and the research topic being addressed.
Taxonomic classification
Taxonomic classification in metagenomics is identifying and classifying microorganisms in a complex microbial community based on DNA sequences. To identify and classify the microorganisms present in the sample, the sequencing data collected from metagenomic samples is compared to existing reference databases, such as the NCBI GenBank.
Many phases are usually involved in the taxonomy classification process. Initially, low-quality reads, adaptor sequences, and host DNA are removed from the raw sequencing data. The remaining reads are then assembled into contigs or scaffolds, which are larger sequences representing segments of microbial genomes. These contigs or scaffolds are then compared to reference databases using bioinformatics tools such as BLAST, DIAMOND, or Kraken to assign them to taxonomic groups based on sequence similarity.
Functional analysis
Based on the DNA sequences of the metagenome, functional analysis in metagenomics entails identifying and describing the functional genes and pathways present in a microbial community. This approach provides insights into the metabolic and biochemical processes occurring within the microbial community, which can aid researchers in understanding the functions that various microorganisms play in the environment.
Typically, functional analysis in metagenomics entails many steps. Initially, low-quality reads, adaptor sequences, and host DNA are removed from the raw sequencing data. The remaining reads are then annotated using bioinformatics techniques like BLAST, Hidden Markov Models (HMMs), or functional gene databases like KEGG or COG.
Data interpretation and visualization
Data interpretation and visualization are essential components of metagenomics research because they allow researchers to acquire insights into the structure and function of microbial communities and successfully communicate their findings to others. Taxonomic profiling, functional profiling, network analysis, 3D visualization, and machine learning are several tools and techniques available for data interpretation and visualization in metagenomics.
Successful metagenomics data interpretation and visualization necessitate not just the use of relevant technologies, but also a comprehension of the underlying biology and statistical principles. As a result, it is critical to interact with professionals in the field and stay current on the newest discoveries in metagenomics research.
Validation and refinement: Validate the results through experimental methods or additional sequencing data, and refine the analysis as needed.
Computational and statistical tools for metagenomic studies:
- Sequence pre-processing: Trimmomatic, Cutadapt
- Sequence assembly: SPAdes, MEGAHIT
- Taxonomic classification: Kraken, MetaPhlAn, DIAMOND
- Functional annotation: HUMAnN, MetaCyc, KEGG
- Statistical analysis: DESeq2, ANCOM
- Visualization: Krona, MEGAN, Cytoscape
- Machine learning: random forests, support vector machines, neural networks
- Cloud computing platforms: Amazon Web Services, Microsoft Azure, Google Cloud Platform
Applications:
As mentioned earlier, metagenomics is a versatile branch of science, having two basic approaches:
Taxonomic application
This method is used to determine the evolutionary relationships of the sequenced gene with microorganism taxonomic groups known in the database. In this example, phylogenetic clusters such as the 16S rRNA gene sequence are targeted, and operational taxonomic units (OTUs) are compared to their amplitude to assess the abundance of microbial species in that specific habitat.
Taxonomic profiling and identification of plant pathogens using next-generation sequencing, in addition to disease diagnostics, microbiome analyses, and outbreak tracing, is one such application of metagenomic analysis. Taxonomic profiling is also used in metabarcoding (similar to metagenomic analysis) to identify all microorganisms, including rare and abundant taxa.
Functional application
This method is used to locate a sequence containing a functional gene with a specific activity or whether the gene is novel with a specific function in a functional pathway. This is accomplished by shotgun metagenomics, which comprises whole-genome sequencing and functional annotation of a gene. Functional annotation is divided into two stages: gene prediction and gene annotation, with gene prediction assisting in the identification of probable protein sequences. Upon identification, the sequences encoding the protein are compared to protein families in databases and functionally annotated by matching the function of the family.
Functional metagenomics widely identifies novel proteins/genes that contribute to the function of the microbial community and influence the environment.
References:
- Thomas T, Gilbert J, Meyer F. Metagenomics – a guide from sampling to data analysis. Microb Inform Exp. 2012 Feb 9;2(1):3.
- Cochran, J.K., Bokuniewicz, H.J. and Yager, P.L., 2019. Encyclopedia of ocean sciences. Academic Press.
- Navgire, G.S., Goel, N., Sawhney, G. et al. Analysis and Interpretation of metagenomics data: an approach. Biol Proced Online 24, 18 (2022). https://doi.org/10.1186/s12575-022-00179-7