Introduction:
Huntington’s disease (HD) is an autosomal, progressive, and dominantly inherited neurodegenerative disorder characterized by choreatic movement impairment and behavioural and mental loss, primarily in the cerebral cortex and striatum.
Dr. George Huntington, a physician who originally reported the disease in a medical paper published in 1872, is named after the disease. Dr. Huntington saw the condition in several generations of a Long Island, New York, family and documented its typical symptoms and course. His precise explanation of the disease aided in raising awareness and comprehension of this previously poorly known and frequently misdiagnosed condition.
A parent with Huntington’s disease (HD) has a 50/50 probability of passing the defective gene to each of their children. More than 200,000 Americans are at risk of inheriting the illness, and there are currently 30,000 Americans with symptoms.
Juvenile Huntington’s disease (JHD) affects children or teenagers in 10% of cases, and it usually progresses more quickly than adult-onset HD.
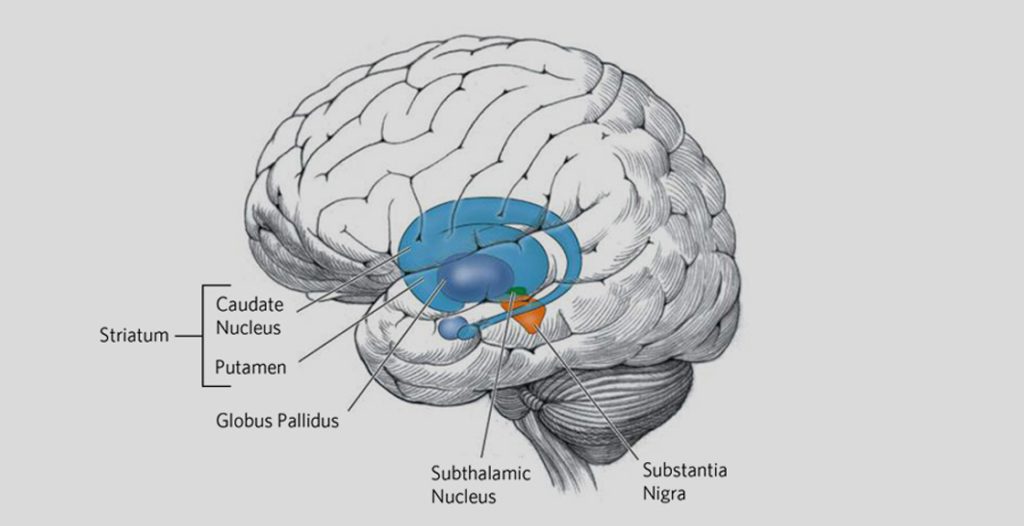
Fig: HD affects the whole brain, but certain areas are more vulnerable than others. Pictured above in blue is the striatum – an area deep in the brain that plays a key role in movement, mood, and behavior control. The striatum is the part of the brain that is most affected by HD.
Epidemiology:
An inherited, rare neurological condition HD is thought to affect 5 to 10 persons per 100,000 people worldwide. The condition can affect people at any age, however it commonly first shows symptoms between the ages of 30 and 50. In some populations, like those with European ancestry, HD is more common; in other populations, like those with African or Asian ancestry, HD is less common. There is some data that suggests that specific geographic areas, such as the Lake Maracaibo region of Venezuela, where the disease affects up to 700 persons per 100,000 people, may have a higher prevalence of HD than others.
Causes:
Huntington’s disease is caused by a mutation in the Huntingtin (htt) gene that is located in chromosome 4, which provides instructions for making a protein called huntingtin that was discovered in 1993. This mutation leads to the production of an abnormal form of the huntingtin protein, which accumulates in nerve cells in the brain and causes damage over time. The mutation that causes Huntington’s disease is inherited in an autosomal dominant pattern, which means that an affected person has a 50% chance of passing the mutation on to each of their children.
In rare cases, Huntington’s disease can also occur spontaneously due to a new mutation in the htt gene, which means that a person can develop the condition even if they have no family history of the disease. However, spontaneous cases are less common than inherited cases.
The specific mechanisms by which the mutant huntingtin protein causes damage to nerve cells in the brain are still being studied, but it is thought to involve a complex interplay of biochemical and cellular processes that disrupt normal brain function over time.
Pathogenesis:
The pathogenic process of HD is explained by various processes such as molecular genetics, which leads to oxidative stress, metabolic malfunctioning, and mitochondrial dysfunction. Huntington’s disease is caused by a complicated chain of events that results in progressive damage to nerve cells in the brain.
A 350 kDa protein known as huntingtin has multiple consensus sequences known as HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1 [target of rapamycin 1]) repeats that are crucial for protein-protein interactions. Huntingtin also contains the polyglutamine sequence at the NH2 terminus. The highest levels of huntingtin expression are found in the neurons of the central nervous system, where it appears to reside mostly in the cytoplasm and be linked to vesicle membranes.
A mutation in the Huntingtin (htt) gene located on chromosome 4 causes the synthesis of an atypical form of the huntingtin protein, which is the primary genetic cause of Huntington’s disease. The mutant protein is likely to disrupt normal cellular functions such as protein breakdown and transport, resulting in the formation of harmful protein aggregates in nerve cells. Toxic aggregates of mutant huntingtin protein can then initiate a series of cellular events, including mitochondrial failure, oxidative stress, inflammation, and changes in calcium signalling and other biochemical processes, all of which can lead to nerve cell death.
Huntington’s disease mostly affects parts of the brain involved in movement control, such as the striatum and cerebral cortex. Individuals with Huntington’s disease may have a variety of motor, cognitive, and mental symptoms as certain parts of the brain deteriorate, including involuntary movements, trouble with coordination and balance, cognitive decline, mood swings, and behavioural changes.
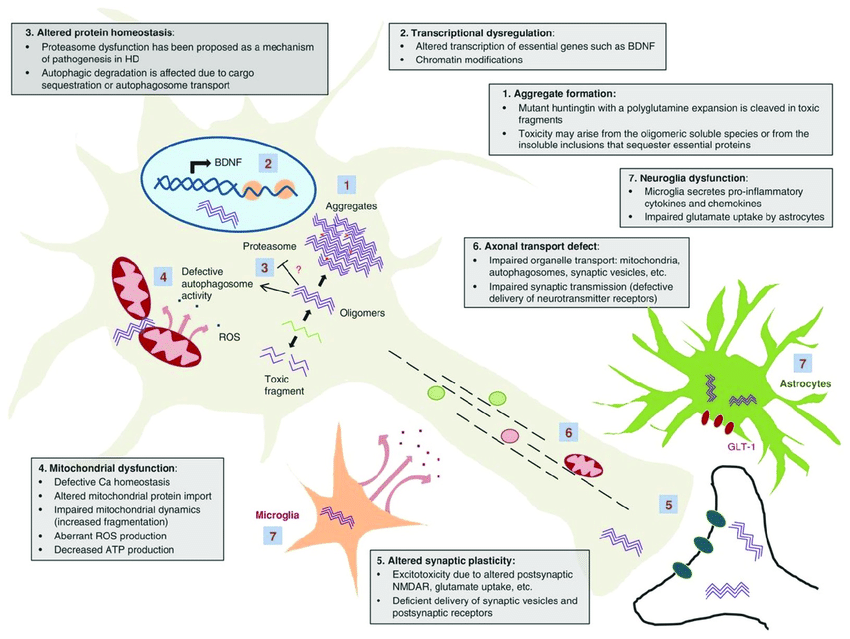
Fig: Schematic of selected mechanisms of pathogenesis in Huntington’s disease
HD is a progressive neurological disorder that is initiated by significant alterations in the HD protein huntingtin (htt). A mutation in htt occurs as a result of an extension inside the CAG repeat tract, resulting in greater lengths of polyglutamine (polyQ) in the encoded protein. In unaffected groups, wild-type alleles have up to 35 CAG repetitions, whereas HD alleles have 36 or more CAG repetitions.
The total number of CAG repeats and the phase of onset of symptoms are inversely related, i.e., bigger CAG repeat extensions are associated with an earlier phase of onset. The htt gene encodes a 348-kDa multidomain protein with a polymorphism glutamine/proline rich domain at the amino terminus.
HD is currently the most widely reported hereditary neurodegenerative disease with diagnostic and predictive genetic testing, and gene-targeted therapy may be available in the near future. One of the first disorders to be genetically tested before birth was HD. Using specialized facilities and genetic testing processes, neuroimaging techniques have certainly given predictive and diagnostic genetic screening for illness diagnosis and its repercussions on sick people and families.
Symptoms:
HD symptoms typically start to show up around middle age. The signs of juvenile Huntington’s disease (JHD) however, appear in early childhood. Children may exhibit early symptoms such as stiffness and seizures in addition to the adult disease’s symptoms. The condition is typically passed along to children with JHD from their fathers.
An autosomal dominant ailment called Huntington’s disease (HD) is characterized by mobility abnormalities and cognitive deterioration. Chorea and loss of coordination are frequent symptoms of motor abnormalities. Depression, psychosis, and obsessive-compulsive disorder are just a few of the psychiatric symptoms that are frequently seen in HD and are very upsetting for the patients.
| Stage | Symptoms |
| Pre-symptomatic | No signs or symptoms are present. A person may have the HD gene mutation but does not show any physical or cognitive changes. |
| Early stage | Mild motor symptoms are noticeable, such as difficulty with balance, coordination, and involuntary movements. Cognitive changes may also be present, including forgetfulness, difficulty with planning, and mood swings. A specific type of movement disorder called chorea is characterized by fleeting, uncontrollable, irregular muscular movements that seem unpredictable and senseless. It is a typical sign of Huntington’s disease (HD), a genetic neurodegenerative condition that affects the brain and causes progressive motor and cognitive function loss. Actually, chorea is frequently among the initial signs of HD and may be present at the start of the condition. |
| Middle stage | Moderate motor symptoms become more apparent, including difficulty with walking and speaking. A person may also experience more cognitive and emotional changes, such as difficulty with decision-making, depression, and irritability. |
| Late stage | Severe motor symptoms are present, and a person may become completely dependent on others for assistance with daily activities, such as eating, dressing, and toileting. Cognitive changes can be profound, with significant difficulty with communication and memory. Behavioural changes, such as aggression and apathy, may also be present. |
| End-stage | The most severe stage of HD, a person requires round-the-clock care and may be bedridden. The ability to communicate is severely compromised, and a person may experience a complete loss of mobility. |
Diagnosis:
Clinical assessment, family history, and genetic testing are all employed to confirm the existence of Huntington’s disease (HD).
Clinical evaluation
To evaluate the patient’s motor, cognitive, and psychological function, a neurologist or other medical professional perform a comprehensive neurological examination. In addition to memory loss, depression, anxiety, irritability, and apathy, the doctor will constantly observe other cognitive and psychiatric signs of HD, such as chorea (involuntary movements). A diagnosis of HD may be suggested by the presence of these symptoms along with a family history of the condition.
A preliminary diagnosis of Huntington’s disease is based primarily on
Neurological examination (Motor symptoms, Sensory symptoms and Psychiatric symptoms)
A neurological exam will assess the patient’s motor function, including gait, coordination, and muscle tone.
Neuropsychological testing (Memory reasoning, mental agility, language skills, Spatial reasoning)
HD can also cause a range of psychiatric symptoms, including depression, anxiety, irritability, and apathy. The healthcare provider will evaluate the patient’s psychiatric function, including mood, affect, and behaviour. The healthcare provider may use standardized tests to evaluate psychiatric symptoms, such as the Hamilton Depression Rating Scale (HAM-D) or the Beck Anxiety Inventory (BAI).
Brain-imaging (Computed tomography (CT), Electroencephalography (EEG), and Magnetic Resonance Imaging (MRI)
Imaging studies may be used to evaluate the brain structure and identify any abnormalities.
Psychiatric assessment
Family history
Since that HD is a hereditary disorder, a family history of the condition must be considered when making a diagnosis. The patient’s family history, including any known occurrences of HD or similar illnesses, will be questioned by the healthcare professional.
Genetic testing
In the last 30 years, direct genetic testing has advanced from the identification of genetic markers that are associated to Huntington’s disease to the identification of linked genetic markers. The diagnosis of HD is confirmed by genetic testing. The genetic test looks for the presence of the mutated huntingtin gene that causes HD. A blood sample or other tissue sample is collected, and the DNA is analysed to determine if the patient has the genetic mutation associated with HD.
The HD gene was finally identified ten years after the initial marker was discovered (Huntington’s Disease Collaborative Research Group, 1993). The IT-15 gene, which encodes the huntingtin protein, is the name of the gene. The aberrant HD gene has an enlarged and unstable DNA region made up of the trinucleotide cytosine-adenine-guanine (CAG) repeats several times in a series. Direct identification of the HD-causing CAG repeat expansion using DNA isolated from peripheral blood samples. CAG repeats are commonly determined using polymerase chain reaction (PCR) and fragment analysis by capillary electrophoresis. HD genetic testing is used to make diagnostic, predictive, and prenatal or preimplantation genetic diagnoses.
| CAG Repeat Size | Interpretation |
| < 26 | Normal, not associated with HD |
| 27 – 35 | Intermediate, not diagnostic for HD but may expand to pathological range in subsequent generations |
| 36 – 39 | Reduced penetrance, may or may not develop HD |
| 40 or more | Pathological range, associated with HD |
The number of CAG trinucleotide repeats in the huntingtin gene on chromosome 4 is referred to as the CAG repeat size in HD. Normal people have less than 26 CAG repeats, whereas people with 40 or more CAG repeats are almost certain to acquire HD. Nevertheless, interpreting CAG repeat sizes in the intermediate (27-35) or low genetic risk range (36-39) is more difficult and may necessitate extra testing or evaluation. It is important to remember that the amount of CAG repeats cannot predict the age of onset or the severity of symptoms in HD. Other factors, including as genetic modifiers, environmental influences, and individual variability, can all have an impact on the disease’s progression.
Treatments:
- It is crucial to remember that treatment for HD is personalized and may differ depending on each individual’s distinct symptoms and needs. Treatment decisions should be made in consultation with a healthcare provider or specialist who is familiar with the treatment of HD. Those with a family history of HD may also benefit from genetic counseling and testing to better understand their risk and prepare for proper medical care and management.
- Physical therapy and rehabilitation, speech therapy and supportive care are the several treatments available that can help manage symptoms and improve quality of life for individuals with HD
- A drug currently approved for tardive dyskinesia (TD) is also effective at treating Huntington’s disease (HD)–associated chorea, a movement disorder that affects most patients with HD, new phase 3 trial results show.
- For adult patients with tardive dyskinesia (TD) and chorea associated with Huntington disease, the FDA has approved extended-release deutetrabenazine (Austedo XR; Teva Pharmaceutical Industries Ltd.) tablets as a once-daily formulation of the presently marketed twice-daily Austedo (HD).
- The FDA has approved deutetrabenazine as the first and only VMAT2 inhibitor for the treatment of TD in adults and chorea associated with HD.
Prevention:
Huntington’s disease (HD) is a hereditary genetic ailment caused by a mutation in the huntingtin gene, and there is currently no known method of preventing it. Genetic testing and counseling, on the other hand, can be useful in identifying an individual’s chance of inheriting the mutant gene and making educated decisions regarding family planning and medical management.
It is crucial to highlight that these changes in lifestyle are not a treatment or a means to avoid HD, but they may assist improve overall health and well-being and maybe delay the onset of symptoms. Moreover, genetic counseling and testing may be recommended for persons who are at risk of inheriting the HD gene mutation in order to better understand their risk and plan for proper medical care and management.
References:
- Roos, R.A. Huntington’s disease: a clinical review. Orphanet J Rare Dis 5, 40 (2010).
- Irfan, Z., Khanam, S., Karmakar, V., Firdous, S.M., El Khier, B.S.I.A., Khan, I., Rehman, M.U. and Khan, A., 2022. Pathogenesis of Huntington’s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sciences, 12(10), p.1389.
- Tassicker RJ, Marshall PK, Liebeck TA, et al. Predictive and pre-natal testing for Huntington disease in Australia: results and challenges encountered during a 10-year period (1994-2003). Clin Genet. Dec 2006;70(6):480-489.
- Jimenez-Sanchez, M., Licitra, F., Underwood, B.R. and Rubinsztein, D.C., 2017. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harbor perspectives in medicine, 7(7), p.a024240.
- Kay C, Hayden MR, Leavitt BR. Epidemiology of Huntington disease. Handb Clin Neurol. 2017; 144:31-46.
- www.hdsa.org/research